一种化合物的结晶形式的制作方法
一种化合物的结晶形式
本技术是申请号为201680029127.3,申请日为2016年5月19日,申请人为爱杜西亚药品有限公司,发明创造名称为“化合物(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的结晶形式”的发明专利申请的分案申请。
技术领域
[0001]
本发明涉及化合物(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇(该化合物在下文中亦称为“化合物”)的结晶形式。
背景技术:
[0002]
(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的制备及其医药用途系阐述于公开的pct申请案wo 2011/007324及wo 2013/175397中。(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇亦可如以下实例1中所阐述来制备。
技术实现要素:
[0003]
本发明的目标系提供化合物的结晶形式且具体而言具有有利的性质的结晶形式。此等有利的性质可包括较高熔点、较佳流动性质、较高热力学稳定性、较低吸湿性、不同溶解性、较高纯度、制造中的较佳再现性(例如较佳过滤参数及较佳固体形成的再现性)、明确形态学及/或较佳长期稳定性。现已发现如本文所述化合物的结晶形式a具有有利的性质。附图简要说明
[0004]
图1显示呈结晶形式a的化合物的x-射线粉末衍射图,其中x-射线粉末衍射图系针对cu kα1辐射展示。图中折射角2θ绘制于水平轴上且计数绘制于垂直轴上。x-射线衍射图显示,在所指示的折射角2θ处具有相较于图中最强峰的以下百分比的相对强度(相对峰强度于括号中给出)的峰(报告在3-30
°
2θ范围内具有大于10%的相对强度的经选择峰):4.0
°
(18%)、4.2
°
(46%)、5.4
°
(100%)、8.0
°
(59%)、8.5
°
(68%)、9.1
°
(12%)、10.8
°
(72%)、12.7
°
(31%)、13.4
°
(18%)、13.6
°
(15%)、14.4
°
(28%)、16.0
°
(18%)、17.0
°
(31%)、17.3
°
(15%)、17.7
°
(22%)、19.0
°
(15%)、19.3
°
(17%)、20.4
°
(47%)、21.0
°
(22%)、21.3
°
(26%)、21.8
°
(22%)、22.8
°
(23%)、25.0
°
(20%)及25.5
°
(19%)。
[0005]
图2显示呈结晶形式b的化合物的x-射线粉末衍射图,其中x-射线粉末衍射图系针对cu kα1辐射展示。图中折射角2θ绘制于水平轴上且计数绘制于垂直轴上。x-射线衍射图显示在所指示折射角2θ处具有相较于图中最强峰的以下百分比的相对强度(相对峰强度在括号中给出)的极宽峰(报告3-30
°
2θ范围内具有大于10%的相对强度的经选择峰):5.9
°
(74%)、7.1
°
(70%)、8.1
°
(35%)、11.9
°
(61%)、14.6
°
(48%)、20.1
°
(65%)及21.5
°
(100%)。
[0006]
图3显示呈结晶形式c的化合物的x-射线粉末衍射图,其中x-射线粉末衍射图系针对cu kα1辐射展示。图中折射角2θ绘制于水平轴上且计数绘制于垂直轴上。x-射线衍射图显示在所指示的折射角2θ处具有相较于图中的最强峰的以下百分比的相对强度(相对峰强度在括号中给出)的峰(报告3-30
°
2θ范围内具有大于10%的相对强度的经选择峰):3.7
°
(11%)、6.4
°
(55%)、7.4
°
(100%)、9.8
°
(77%)、12.8
°
(49%)、13.2
°
(28%)、14.7
°
(15%)、17.0
°
(24%)、19.5
°
(24%)、20.5
°
(22%)、21.2
°
(19%)、23.3
°
(17%)及25.9
°
(20%)。
[0007]
图4显示呈非晶形状态的化合物的x-射线粉末衍射图,其中x-射线粉末衍射图系针对cu kα1辐射展示。图中折射角2θ绘制于水平轴上且计数绘制于垂直轴上。x-射线衍射显示如针对非晶形材料所获得的典型图。
[0008]
为避免任何疑问,上文所列示的峰阐述图1至3中所示的x-射线粉末衍射的实验结果。应理解,与上文峰清单相比,仅需要选择特征峰以充分且明确表征本发明呈各别结晶形式的化合物。
具体实施方式
[0009]
1)本发明的第一实施例系关于化合物(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的结晶形式(例如基本上纯结晶形式),其特征在于在x-射线粉末衍射图中在以下折射角2θ处存在峰:5.4
°
、8.5
°
及10.8
°
。
[0010]
2)在另一实施例中,本发明系关于如实施例1)的结晶形式,其特征在于在x-射线粉末衍射图中在以下折射角2θ处存在峰:4.2
°
、5.4
°
、8.0
°
、8.5
°
及10.8
°
。
[0011]
3)在另一实施例中,本发明系关于如实施例1)的结晶形式,其特征在于在x-射线粉末衍射图中在以下折射角2θ处存在峰:4.2
°
、5.4
°
、8.0
°
、8.5
°
、10.8
°
、12.7
°
、14.4
°
、17.7
°
、20.4
°
及21.3
°
。
[0012]
4)在另一实施例中,本发明系关于如实施例1)至3)中任一项的结晶形式,其基本上显示如图1中所绘示的x-射线粉末衍射图案。
[0013]
5)在另一实施例中,本发明系关于如实施例1)至4)中任一项的结晶形式,其具有如藉由差示扫描量热法使用如本文所述方法来测定的约79℃的熔点。
[0014]
6)在另一实施例中,本发明系关于化合物(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的结晶形式(例如基本上纯结晶形式),其可藉由以下获得:i)将20mg呈非晶型的(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇溶解于0.1ml乙酸乙酯中;ii)历经1h逐渐添加0.9ml正庚烷;及iii)使其在20-25℃下封闭静置过夜;或iv)将25-30mg呈非晶型的(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇与5ml乙酸乙酯/正庚烷1/9(体积/体积)混合并加热至70℃;及v)使溶液冷却至20-25℃且将其在4℃下储存过夜。7)在另一实施例中,本发明系关于如实施例6)的结晶形式,其特征在于在x-射线
粉末衍射图中在以下折射角2θ处存在峰:5.4
°
、8.5
°
及10.8
°
。8)在另一实施例中,本发明系关于如实施例6)的结晶形式,其特征在于在x-射线粉末衍射图中在以下折射角2θ处存在峰:4.2
°
、5.4
°
、8.0
°
、8.5
°
及10.8
°
。9)在另一实施例中,本发明系关于如实施例6)的结晶形式,其特征在于存在x-射线粉末衍射图中在以下折射角2θ处在峰:4.2
°
、5.4
°
、8.0
°
、8.5
°
、10.8
°
、12.7
°
、14.4
°
、17.7
°
、20.4
°
及21.3
°
。10)在另一实施例中,本发明系关于如实施例6)的结晶形式,其基本上显示如图1中所绘示的x-射线粉末衍射图案。11)在另一实施例中,本发明系关于如实施例6)至10)中任一项的结晶形式,其具有如藉由差示扫描量热法使用如本文所述的方法所测定的约79℃的熔点。12)在另一实施例中,本发明系关于如实施例1)至5)中任一项的结晶形式,其可藉由实施例6)的方法获得。基于如上文中所揭示的不同实施例1)至12)的依赖性,以下实施例由此系可能的且意欲呈个别化形式且以个别化形式明确揭示于此:1、2+1、3+1、4+1、4+2+1、4+3+1、5+1、5+2+1、5+3+1、5+4+1、5+4+2+1、5+4+3+1、6、7+6、8+6、9+6、10+6、11+6、11+7+6、11+8+6、11+9+6及11+10+6。同样,实施例12)系关于以个别化形式明确揭示于此的以下实施例:可藉由6获得的1、可藉由6获得的2+1、可藉由6获得的3+1、可藉由6获得的4+1、可藉由6获得的4+2+1、可藉由6获得的4+3+1、可藉由6获得的5+1、可藉由6获得的5+2+1、可藉由6获得的5+3+1、可藉由6获得的5+4+1、可藉由6获得的5+4+2+1及可藉由6获得的5+4+3+1。在上述列表中,数字系指根据上文所提供其编号的实施例,而“+”指示与另一实施例的依赖性。不同个别化实施例由顿号隔开。换言之,例如,“5+4+1”系指实施例5)依赖于实施例4),该实施例4)依赖于实施例1),即,实施例“5+4+1”对应于特征进一步在于实施例4)及5)的特征的实施例1)。此外,“可藉由6获得”意指所指示的个别化实施例可藉由实施例6)的方法获得。除非另外明确阐述的定义提供更广或更窄的定义,否则本文所提供的定义意欲一致地适用于如实施例1)至12)中任一项及(已作必要的修正)贯穿整个说明及申请专利范围中所定义的标的物。应充分理解,术语或表述的定义或较佳定义独立于(及与的组合)如本文所定义任一或所有其他术语或表述的任一定义或较佳定义来定义且可替代各别术语或表述。术语“基本上纯”在本发明的上下文中应理解为尤其意指结晶形式a中存在至少90、较佳至少95且最佳至少99重量%的(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇。当定义(例如)x-射线粉末衍射图中峰的存在时,常见方式系藉助s/n比率(s=信号,n=噪声)来进行。根据此定义,当阐述x-射线粉末衍射图中必须存在一峰时,应理解x-射线粉末衍射图中的该峰的定义为s/n比率大于x(x为大于1的数值),通常大于2,尤其大于3。在阐述结晶形式基本上显示如图1中所绘示的x-射线粉末衍射图案的情况下,术语“基本上”意指该图中所绘示图形至少必须存在主要峰,亦即彼等相较于图中最强峰的相
对强度大于10%、尤其大于20%的峰。然而,熟习x-射线粉末衍射的本领域技术人员将认识到,x-射线粉末衍射图中的相对强度可能由于较佳取向效应而产生较强的强度变化。除非使用时涉及温度,否则在本技术案中置于数值“x”前的术语“约”系指自x-x的10%延伸至x+x的10%的区间,且较佳系指自x-x的5%延伸至x+x的5%的区间。在温度的特定情形下,在本技术案中置于温度“y”前的术语“约”系指自温度y-5℃延伸至y+5℃的区间,且较佳系指自y-3℃延伸至y+3℃的区间。当在本技术案中指定峰的衍射角2西塔(2θ)时,应理解所给出的值应理解为自该值-0.2
°
至该值+0.2
°
且较佳自该值-0.1
°
至该值+0.1
°
的区间。本发明结晶形式a可用作药物,例如呈用于经肠或非经肠投与(例如尤其经口投与)的医药组合物形式,且适用于减少循环淋巴细胞的数目且用于预防及/或治疗哺乳动物(例如尤其人类)中与活化的免疫系统相关的疾病或病症。医药组合物的制造可根据本领域技术人员熟悉的方式(参见例如remington,the science and practice of pharmacy,第21版(2005),第5部分,“pharmaceutical manufacturing”[由lippincott williams&wilkins出版])藉由将本发明结晶形式a(可选地与其他有治疗价值的物质组合)连同适宜无毒惰性医药上可接受的固体或液体载体材料及(若需要时)常用医药佐剂制成盖仑制剂(galenical)投与形式来达成。化合物的结晶形式a可呈单一组份使用或呈与化合物的其他结晶形式或非晶型的混合物使用。可使用本发明结晶形式a来治疗及/或预防的与活化的免疫系统相关的疾病或病症系阐述于(例如)wo 2011/007324中。欲使用本发明结晶形式a来治疗及/或预防的较佳疾病或病症选自由以下组成的群:移植器官(例如肾、肝、心脏、肺、胰脏、角膜及皮肤)排斥;移植物抗宿主疾病;自体免疫症候群,包括斯耶格伦氏综合征(syndrome)、脊椎关节病/僵直性脊椎炎、幼年型关节炎、亚急性皮肤狼疮、盘状红斑狼疮、狼疮性肾炎、全身性硬化、弥漫性皮肤全身性硬化、血管炎(例如韦格纳(m.wegener))、巨细胞动脉炎、贝切特氏病(behcet disease)、非传染性眼色素层炎、古巴士德氏症候群(goodpasture syndrome)、葛瑞夫兹氏病(grave's disease)、格林-巴利症候群(guillain barr
éꢀ
syndrome)、原发性胆汁性肝硬化、原发性硬化性胆管炎、自体免疫肝炎、多发性肌炎、皮肌炎、微小性结肠炎、乳糜泻、类肉瘤病、白斑症、斑秃、慢性发炎性去髓鞘型多发性神经病变(cidp)、拉斯马森脑炎(rasmussen's encephalitis)、类风湿性关节炎、多发性硬化、诸如克罗恩氏病(crohn's disease)及溃疡性结肠炎等发炎性肠病、牛皮癣、牛皮癣关节炎、诸如桥本氏甲状腺炎(hashimoto’s thyroiditis)等甲状腺炎、眼色素层视网膜炎及全身性红斑狼疮;异位性疾病,例如鼻炎、结膜炎及异位性皮肤炎;气喘;i型糖尿病;及包括风湿热的感染后自体免疫疾病。极佳地,本发明结晶形式a用于治疗全身性红斑狼疮。本发明亦系关于用于预防或治疗本文所提及或wo 2011/007324中所提及的疾病或病症的方法,其包含将医药活性量的本发明结晶形式a投与个体(尤其人类个体)。此外,本发明结晶形式a亦与一或若干种免疫调节剂组合以用于预防及/或治疗本文所提及的疾病及病症。根据本发明的较佳实施例,该等试剂选自由以下组成的群:免疫抑制剂、皮质类固醇、非类固醇抗发炎药物、细胞毒性药物、分子黏附抑制剂、细胞介素、细胞
介素抑制剂、细胞介素受体拮抗剂及重组细胞介素受体。本发明亦系关于本发明结晶形式a的用途,其用于制备可选地与一或若干种免疫调节剂组合用于预防或治疗本文所提及或wo 2011/007324中所提及的疾病及病症的医药组合物。(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇可(例如)如公开的pct申请案wo 2011/007324(具体而言参见实例2)中所阐述或藉由使用如公开的pct申请案wo 2013/175397中所揭示的制备方法来制备。具体而言,化合物亦可如下文所述来制备。实验部分以下实例更详细地阐释本发明。温度系以摄氏度给出。若未另外阐明,则室温系在18-25℃范围内且百分比系以重量计来给出。如本文所用缩写:a/a
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
面积/面积api
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
活性医药成份ca.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
约dcm
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二氯甲烷dipea
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀhü
ning碱,二乙基异丙胺dmf
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲基甲酰胺dmso
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
二甲亚砜dsc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
差示扫描量热法eq.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
当量etoac
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙酸乙酯etoh
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
乙醇fig.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
图h
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
小时1h-nmr
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
氢-1核磁共振hplc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
高效液相层析hpmc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
羟基丙基甲基纤维素lc-ms
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
液相层析-质谱meoh
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
甲醇min
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
分钟m.p.
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
熔点rh
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
相对湿度rt
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
室温tbtu
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
四氟硼酸2-(1h-苯并三唑-1-基)-1,2,3,3-四甲基脲鎓盐tea
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
三乙胺tfa
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
三氟乙酸thf
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
四氢呋喃tlc
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
薄层层析
trꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
滞留时间xrpd
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
x-射线粉末衍射所用方法:1h-nmr400mhz,bruker;化学位移系相对于所用溶剂以ppm给出。x-射线粉末衍射分析x-射线粉末衍射图案系于配备有使用cuka辐射以反射模式(偶合2θ/θ)操作的lynxeye检测器的bruker d8 advance x-射线衍射仪上收集。通常,x-射线管在40kv/40ma下运行。施加在3-50
°
2θ的扫描范围内的0.02
°
(2θ)的步长及76.8秒的步时。发散狭缝设定为固定的0.3。以0.5mm的深度将粉末轻微压至硅单晶样品架中且在量测期间使试样在其自身平面上旋转。在使用仪器评估软件(eva)去除kα2组份之后,使用cu kα1来报告衍射数据。如本文所提供的2θ值的准确度在+/-0.1-0.2
°
范围内,如通常记录的x-射线粉末衍射图案通常存在的情形。差示扫描量热法(dsc)于配备有34位自动取样器的mettler toledo stare系统(dsc822e模块,具有陶瓷传感器及9.20版star软件的量测单元)上收集dsc数据。使用标准铟对仪器的能量及温度进行校准。通常将1-5mg的各试样在自动穿刺的铝盘中以10℃min-1
(除非另外阐明)自-20℃加热至280℃。在试样上方维持20ml min-1
的氮吹扫。峰温度系针对熔点报告。实例1(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的制备a)(r)-n-((2-环戊基-6-甲氧基异烟碱酰基)氧基)-4-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-乙基-5-甲基苯甲脒向30l反应器中添加2-环戊基-6-甲氧基-异烟碱酸(1.27kg,1当量;可(例如)如wo 2013/175397中所阐述来制备)、dmf(17ml)及dcm(18l)。在20℃下经30min向悬浮液中添加草酰氯(534ml,1.1当量)。将混合物搅拌30min。藉由lc-ms分析来确认反应完全。在《30℃下经20min的时段将(r)-4-(2,2-二甲基-[1,3]二氧戊环-4-基甲氧基)-3-乙基-n-羟基-5-甲基-苯甲脒(1.77kg,1当量;可如wo 2011/007324中所述来制备)及tea(1.78l,2.2当量)于dcm(4l)中的溶液添加至酰基氯中。在搅拌15min后,藉由lc-ms分析来确认反应完全。用水(7l)洗涤反应混合物。在55℃及减压下移除溶剂(18l)。添加etoh(26l),将悬浮液冷却至0℃并过滤。用etoh(7l)洗涤滤饼。将固体在50℃下于旋转蒸发器上干燥以获得灰白色固体。产率:2261g(77%)。lc-ms:纯度:100%a/a,tr=1.886min,[m+1]
+
=512;1h-nmr(cdcl3):δ7.43(s,2h),7.34(s,1h),7.12(s,1h),5.16(s,2h),4.52(quint,j=5.8hz,1h),4.21(dd,j1=8.3hz,j2=6.9hz,1h),3.98(s,3h),3.96(m,1h),3.83(m,2h),3.19(m,1h),2.70(m,2h),2.33(s,3h),2.06(m,2h),1.85(m,4h),1.71(m,2h),1.46(d,j=21.3hz,6h),1.25(t,j=7.6hz,3h)。b)(r)-5-(2-环戊基-6-甲氧基吡啶-4-基)-3-(4-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-乙基-5-甲基苯基)-1,2,4-噁二唑将(r)-n-((2-环戊基-6-甲氧基异烟碱酰基)氧基)-4-((2,2-二甲基-1,3-二氧戊
环-4-基)甲氧基)-3-乙基-5-甲基苯甲脒(2150g,1当量)于甲苯(10l)中的混合物加热回流4h。将水收集于dean stark装置中。将溶液在70℃及减压下浓缩至干燥以获得黄色油。产率:2116g(102%)。lc-ms:纯度:96%a/a(4%a/a甲苯),tr=2.665min,[m+1]
+
=494;1h-nmr(cdcl3):δ7.87(d,j=6.3hz,2h),7.50(s,1h),7.30(s,1h),4.55(quint,j=5.8hz,1h),4.23(dd,j1=8.4hz,j2=6.5hz,1h),4.01(m,4h),3.90(m,2h),3.24(m,1h),2.77(m,2h),2.40(s,3h),2.09(m,2h),1.88(m,4h),1.73(m,2h),1.50(s,3h),1.48(d,j=22.0hz,6h),1.32(t,j=7.5hz,3h)。用于步骤a)及b)中的lc-ms方法:agilent g1956b(ms,电离:esi+,apci),agilent g1312b bin pump,agilent g1315c dad,agilent g1316b(恒温管柱隔室),agilent g1367c(自动取样器)。注射体积:2μl;管柱:kinetex c18,2.6μm,2.1
×
50mm;温度:40℃;流速:1ml/min;梯度:水/乙腈:2.8min内95:5至5:95,然后0.2min内95:5。c)(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇向30l b
ü
chi反应器中添加(r)-5-(2-环戊基-6-甲氧基吡啶-4-基)-3-(4-((2,2-二甲基-1,3-二氧戊环-4-基)甲氧基)-3-乙基-5-甲基苯基)-1,2,4-噁二唑(2.28kg,1当量)及etoh(5l)。将溶液加热至45℃且添加1n hcl(3l,0.75当量)。将所得混合物在45℃下搅拌1h且在减压(400毫巴)下再搅拌3h。将混合物用32%naoh(300ml,0.75当量)中和且在60℃及减压下浓缩直至达到最小搅拌体积(约2l)为止。将反应器用氮气设定为正常压力。用etoac(20l)稀释残留物。用水(2
×
10l)洗涤混合物。将有机层在60℃及减压下浓缩以获得黄色油。产率:2053g(98%)。产生第二批;产率:1907g(98%)。结晶:在pyrex烧瓶中将两批(2053g+1907g)合并并溶解于etoac(5.5l)中(api溶液)。向30l反应器中添加呈结晶形式a的(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇(14g)及正庚烷(30l)。将悬浮液加热至40℃且在40℃下经1h的时段添加api溶液。将悬浮液再搅拌0.5h,冷却至20℃且经30l b
ü
chi吸滤器过滤。用正庚烷(6l)洗涤产物。将产物于施加轻柔氮气流的吸滤器上干燥2天。产率:3300g(83%),纯度(hplc方法):99.51%a/a;熔点:约79℃(dsc),呈结晶形式a的化合物(图1)。1h-nmr(d
6 dmso):δ7.78(s,2h),7.53(s,1h),7.26(s,1h),4.98(d,j=4.6hz,1h),4.65(s,1h),3.94(s,3h),3.86(m,2h),3.75(m,1h),3.50(t,j=5.4hz,2h),3.28(m,1h),2.75(d,j=7.5hz,2h),2.35(s,3h),2.03(m,2h),1.81(m,4h),1.69(m,2h),1.22(t,j=7.5hz,3h)。hplc方法:hplc系统agilent 1100;注射体积:5μl;管柱:zorbax eclipse xdb c18,3.5μm,150mm
×
4.6mm;温度:30℃;流速:1ml/min;检测波长:250nm;梯度:水/乙腈:2.8min内95:5至5:95,然后0.2min内95:5。溶析剂:溶析剂a:水/meoh/tfa(95/5/0.05),溶析剂b:水/meoh/tfa(5/95/0.05);梯度:0-1min 40%a,7-22min 0%a,22.1-27min 40%a。
非晶形化合物的制备:非晶形化合物可藉由针对公开的pct申请案wo 2011/007324的实例2所阐述的方法获得。该方法系如下:a)向2-环戊基-6-甲氧基-异烟碱酸(162mg,0.732mmol)于dmf(2ml)及thf(10ml)中的溶液中添加dipea(189mg,251μl,1.46mmol),随后添加tbtu(235mg,0.732mmol)。将混合物在室温下搅拌10min,然后添加(r)-4-(2,2-二甲基-[1,3]二氧戊环-4-基甲氧基)-3-乙基-n-羟基-5-甲基-苯甲脒(226mg,0.732mmol)。将混合物在室温下搅拌1h,然后将其用etoac稀释且用水洗涤。分离有机层并浓缩。将残留物(375mg)溶解于二氧六环(10ml)中并将混合物加热至105℃持续18h。将混合物冷却至室温,浓缩且于制备性tlc板(硅胶,0.5mm)上使用含有10%甲醇的dcm纯化粗产物以获得黄色油形式的4-{3-[4-((r)-2,2-二甲基-[1,3]二氧戊环-4-基甲氧基)-3-乙基-5-甲基-苯基]-[1,2,4]噁二唑-5-基}-2-环戊基-6-甲氧基-吡啶(396mg);lc-ms:tr=1.39min,[m+h]
+
=494.31。b)将4-{3-[4-((r)-2,2-二甲基-[1,3]二氧戊环-4-基甲氧基)-3-乙基-5-甲基-苯基]-[1,2,4]噁二唑-5-基}-2-环戊基-6-甲氧基-吡啶(390mg,790μmol)于4m hcl于二氧六环(16ml)中的溶液在室温下搅拌90min,然后将其浓缩。于制备性tlc板上使用含有10%甲醇的dcm来纯化粗产物以获得灰白色泡沫形式的(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇(80mg);lc-ms:tr=1.20min,[m+h]
+
=454.32;1h-nmr(400mhz,cdcl3):δ7.91(d,j=2.0hz,1h),7.89(d,j=2.0hz,1h),7.53(d,j=0.8hz,1h),7.32(d,j=1.0hz,1h),4.16-4.22(m,1h),4.03(s,3h),3.96-3.99(m,2h),3.93(dd,j1=4.3hz,j2=11.3hz,1h),3.88(dd,j1=5.5hz,j2=11.3hz,1h),3.21-3.31(m,1h),2.79(q,j=7.6hz,2h),2.74(s br,1h),2.43(s,3h),2.07-2.17(m,2h),1.85-1.96(m,4h),1.70-1.81(m,2h),1.34(t,j=7.5hz,3h);xrpd图如图4中所显示。在制备非晶形化合物的上述描述中,化合物系由1h-nmr(bruker avance ii,400mhz ultrashield
tm
,400mhz(1h),100mhz(
13
c);化学位移系以百万分率(ppm)相对于四甲基硅烷(tms)来报告且多重性系以s(单峰)、d(双峰)、t(三重峰)、q(四重峰)、quint(五重峰)、h(六重峰)、hept(七重峰)或m(多重峰)形式给出,br=宽,偶合常数系以hz给出);及/或由lc-ms(finnigan msq
tm plus或msq
tm
测量器(dionex,瑞士)与agilent g4220a泵及agilent g4212a dad(agilent,瑞士),管柱:zorbax rrhd sb-aq,1.8μm,3.0
×
20mm(agilent);梯度:1.2min内5-95%含有0.04%三氟乙酸的水中的乙腈,流速:1.6ml/min;tr系以min给出)来表征。藉由涂布有硅胶60f
254
(0.5mm)的制备性tlc玻璃板来纯化化合物。实例2:形式a的制备将20mg呈非晶型的化合物溶解于0.1ml etoac中且经1h逐渐添加0.9ml正庚烷。在20-25℃下封闭静置过夜后收集所形成的固体且其由呈结晶形式a的化合物组成。或者,将25-30mg呈非晶型的化合物与5ml etoac/正庚烷1/9(体积/体积)混合且加热至70℃。使溶液冷却至20-25℃且然后在4℃下储存过夜。收集所获得固体且该固体系呈结晶形式a的化合物。藉由dsc观察到在约66℃至约88℃范围内的宽的吸热事件,其中峰在约79℃(结晶形式a的熔点)处。
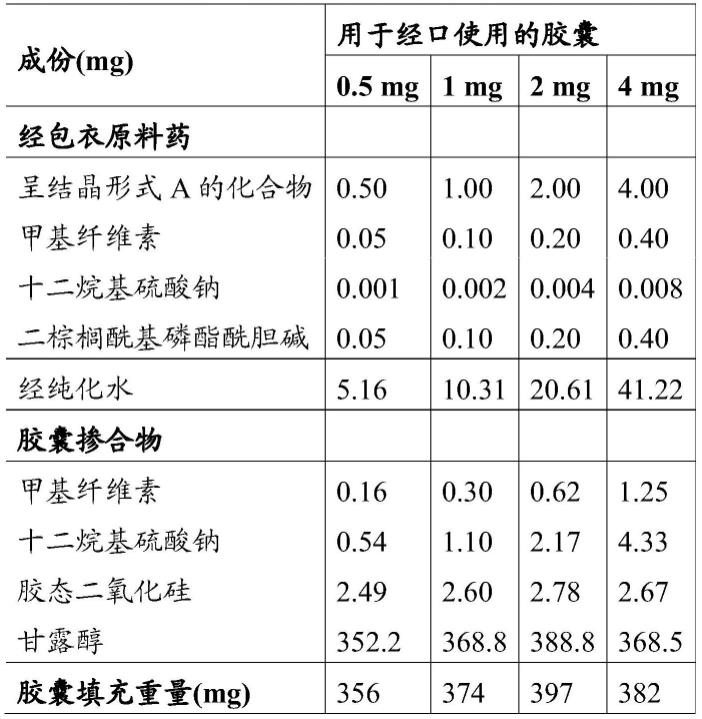
实例3:形式b的制备将0.5g呈结晶形式a的化合物、2.5ml dcm及3ml正庚烷混合并过滤至培养皿(具有约9cm的直径)中。使溶液在20-25℃下蒸发过夜。收集固体残留物且在真空下干燥(2毫巴持续1h)。如此获得的固体残留物系呈结晶形式b的化合物,如图2中所示。藉由dsc观察到在约44℃至约63℃范围内的宽的吸热事件,其中峰在约58℃(结晶形式b的熔点)处。实例4:形式c的制备在15ml褐色玻璃小瓶中将266mg呈结晶形式a的化合物及36mg尿素溶解于10ml甲醇中。将小瓶在20-25℃下敞口静置用以蒸发溶剂。一旦所有溶剂蒸发且最晚在1周后即刻添加10ml水且藉由磁力搅拌在20-25℃下将试样搅拌5天。过滤悬浮液且将回收的固体在2毫巴下干燥1h。如此获得的固体残留物系呈结晶形式c的化合物,如图3中所显示。藉由dsc观察到在约30℃至约60℃范围内的宽的吸热事件,其中峰在约48℃(结晶形式c的熔点)处。实例5:呈结晶形式a、b及c的化合物的吸湿性的比较方法:重量蒸汽吸附(gvs)分析:在于25℃下在以步进模式操作的多试样仪器sps-100n(projekt messtechnik,ulm,德国)上对呈结晶形式a、b及c的化合物同时实施量测。使试样在40%rh下平衡,然后开始施加预定湿度程序(40-0-95-0-95-40%rh,步进为5%δrh且每步最大平衡时间为24小时)。使用约20mg至30mg的各试样。根据欧洲药典技术指南(european pharmacopeia technical guide,1999,第86页)进行吸湿性分类,即,不吸湿:质量增加少于0.2%质量/质量;轻微吸湿:质量增加少于2%且等于或大于0.2%质量/质量;吸湿:质量增加少于15%且等于或大于2%质量/质量。虑及在首次吸附扫描中介于40%相对湿度与80%相对湿度之间的质量变化。形式a:《0.2%质量增加:不吸湿形式b:0.5%质量增加:轻微吸湿形式c:0.8%质量增加:轻微吸湿实例6:含有0.5mg、1mg、2mg及4mg呈结晶形式a的化合物的胶囊
由于活性物质(s)-3-{4-[5-(2-环戊基-6-甲氧基-吡啶-4-基)-[1,2,4]噁二唑-3-基]-2-乙基-6-甲基-苯氧基}-丙烷-1,2-二醇的极低水溶解性(水中约0.06μg/ml)及差的润湿能力,原料药包衣作为第一步骤。在此程度上,将甲基纤维素(methocel
tm a15 premium lv,悬浮剂)、十二烷基硫酸钠及二棕榈酰基磷酯酰胆碱(润湿剂)在搅拌下相继添加至纯化水中:各赋形剂仅在前一者完全溶解后添加。将api(即呈结晶形式a的化合物)经由筛孔大小40筛分,添加至甲基纤维素溶液中并搅拌3h,直至形成完全均匀悬浮液为止。将悬浮液喷雾干燥(出口空气温度40-50℃,干燥气体流速110kg/h,雾化n2气体流速8kg/h,n2雾化压力0.7巴),此产生经包衣api。包衣使得活性物质溶解性增加(水中约29μg/ml)。经包衣后,测定经包衣的原料药中的原料药含量且最后相应地校正用于下一步骤中的材料的量。将经包衣api与甲基纤维素(methocel
tm a15 premium lv)、十二烷基硫酸钠及一部分的甘露醇(部分的甘露醇(m 200甘露醇)一起筛分并混合。在筛分后在2步内将更多份甘露醇添加至掺合物中,每次皆随后进行混合。然后将胶态二氧化硅(200)与其余甘露醇一起筛分并添加至粉末掺合物中。将最终混合物进一步掺和。然后将粉末填充入“0”型白色不透明hpmc胶囊中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1