一种新型1-甲基吡咯烷-2-丙烯酸的制备方法与流程
1.本发明涉及有机合成技术领域,具体涉及一种新型1-甲基吡咯烷-2-丙烯酸的制备方法。
背景技术:
2.1-甲基吡咯烷-2-基丙烯酸盐酸盐结构式为:
[0003][0004]
属于d-脯氨酸衍生物,是恒瑞医药公司研发的抗肿瘤药物新药吡咯替尼 (pyrotinib)的一个关键中间体。
[0005]
目前的合成方法中有以下三种:
[0006]
1、wo2017186140a1中公开方法:
[0007][0008]
该反应需要柱层析纯化,不利于生产。
[0009]
2、cn108314639b中公开的方法:
[0010][0011]
该方法跟wo2017186140a1的方法一样,需要在boc保护下,先将脯氨酸反应成醇,再进行氧化得到醛。
[0012]
3、cn111018767b中公开的方法:
[0013][0014]
该方法制备酰氯的过程中,产物收率较低,且整个反应容易粘稠,后处理不方便,同时在还原过程中,需要特殊催化剂,价格昂贵,成本较高。
技术实现要素:
[0015]
针对现有技术中所存在的不足,本发明的目的在于提供另一种新型1-甲基吡咯烷-2-丙烯酸的制备方法,无需将脯氨酸反应成醇,操作简单,处理方便。
[0016]
为实现上述目的,本发明采用了如下的技术方案:一种新型1-甲基吡咯烷-2-丙烯酸的制备方法,其特征在于:以d-脯氨酸为原料,经过甲基化得到中间体z1或采用boc保护基或cbz保护基对氮进行保护得到boc保护的中间体z1或cbz保护的中间体z1,中间体z1、boc保护的中间体z1或cbz保护的中间体z1与十二硫醇进行酯化反应得到甲酸硫酯中间体,甲酸硫酯中间体再经过福山还原得到1-甲基吡咯烷-2-甲醛或boc保护的或cbz保护的甲醛中间体,所述boc保护的或cbz保护的甲醛中间体再脱去相应的保护基并甲基化得到1-甲基吡咯烷-2-甲醛,反应式为:
[0017][0018]
上述方案中:1-甲基吡咯烷-2-甲醛再经过witting反应得到1-甲基吡咯烷-2-丙烯酸乙酯,1-甲基吡咯烷-2-丙烯酸乙酯经过水解后得到1-甲基吡咯烷-2-丙烯酸或其盐酸盐,反应式为:
[0019][0020]
上述方案中:中间体z1的合成为:将d-脯氨酸溶于甲醇中,加入多聚甲醛和催化剂,投入高压釜中,通入氢气,反应至监控原料反应完全,过滤,滤液减压浓缩至干,得到白色固体z1。
[0021]
上述方案中:boc保护的中间体z1的合成为:在反应容器中投入d-脯氨酸和二氯甲烷,室温搅拌下加入三乙胺,搅拌后,加入二碳酸二叔丁酯,保温25-30℃反应5-6小时,降温至0℃,用盐酸调节ph值1-2,有机层用饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩至干,得无色油状物,加入石油醚打浆,析出白色固体,过滤,干燥得boc保护的中间体z1。
[0022]
上述方案中:cbz保护的中间体z1的合成为:在反应容器中投入d-脯氨酸和水,搅拌溶清,加入碳酸钠,搅拌30分钟,降温至0℃左右,滴加预先配置的氯甲酸苄酯甲苯溶液,滴加完毕,保温0℃左右,反应30分钟,升温至20-25℃反应至反应完毕,加入水,过滤,滤液分层,水层用甲苯提取,合并水层,用盐酸调ph值至1-2,用乙酸乙酯萃取两次,合并有机相,无水硫酸镁干燥,过滤,减压浓缩至干,得到油状物cbz保护的中间体z1。
[0023]
上述方案中:甲酸硫酯中间体的合成为:在反应容器中加入中间体z1或 boc保护的中间体z1或cbz保护的中间体z1,加入缩合剂1-乙基-3(3-二甲基丙胺)碳二亚胺、dmap,加入二氯甲烷,氮气置换后,氮气保护下,降温至 0-5℃;滴加预先配置好的十二硫醇二氯甲烷溶液,保温反应1-1.5小时,加水洗涤,有机层用饱和食盐水洗涤,无水硫酸镁干燥,溶剂旋干,得到黄色油状物甲酸硫酯中间体。
[0024]
上述方案中:1-甲基吡咯烷-2-甲醛的合成为:在反应容器中投入氮甲基保护的甲酸硫酯中间体、pd/c、丙酮,氮气置换后,氮气保护下,降温至0-5℃,滴加三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入二氯甲烷提取,饱和碳酸钠溶液洗涤后,无水硫酸镁干燥,旋干,得1-甲基吡咯烷-2-甲醛。
[0025]
上述方案中:在反应容器中投入boc保护的甲酸硫酯、pd/c、丙酮,氮气置换后,氮气保护下,降温至0-5℃,滴加三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入50ml二氯甲烷提取, 40ml饱和碳酸钠溶液洗涤后,无水硫酸镁干燥后,旋干,得boc保护的甲醛中间体;
[0026]
在反应容器中中投入boc保护的甲醛中间体,加入二氯甲烷,室温搅拌下滴加三氟乙酸,室温反应1-1.5小时,反应完毕,将反应液浓缩至干,加入二氯甲烷,加入碳酸钠,搅拌下,滴加入碘甲烷,室温下搅拌反应2-3小时,过滤,减压浓缩至干,得到1-甲基吡咯烷-2-甲醛。
[0027]
上述方案中:在反应容器中投入cbz保护的甲酸硫酯中间体、pd/c、丙酮,氮气置换后,氮气保护下,降温至0-5℃,滴加三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入50ml二氯甲烷提取,40ml饱和碳酸钠溶液洗涤后,无水硫酸镁干燥后,旋干,得cbz保护的甲醛中间体;
[0028]
在高压釜中投入cbz保护的甲醛中间体、甲醇和多聚甲醛,加入pd/c,通入氢气,50℃反应,监控原料反应完全,过滤,滤液减压浓缩至干,得到 1-甲基吡咯烷-2-甲醛。
[0029]
上述方案中:在反应容器中加入1-甲基吡咯烷-2-甲醛、二氯甲烷, 10~15℃下,
滴加入预先配置好的乙氧甲酰基亚甲基三苯基膦的二氯甲烷溶液,保温反应16-18小时,反应完毕后,加水,用盐酸调节ph值至2-3,分去有机层,水层加入二氯甲烷,用4n氢氧化钠调ph值至8-9,取有机层减压浓缩至干,得到酒红色油状物1-甲基吡咯烷-2-丙烯酸乙酯;
[0030]
在反应容器中加入1-甲基吡咯烷-2-丙烯酸乙酯、加入氢氧化钠溶液, 40℃反应2-3小时,反应完毕,将反应液浓缩至干,用盐酸溶液调节ph值至 6-7,加入乙酸乙酯萃取,萃取完毕,用盐酸调ph值至2-3,将反应液旋干,得到白色固体,异丙醇重结晶,得到产品白色固体1-甲基吡咯烷-2-丙烯酸。
[0031]
有益效果:本发明以d-脯氨酸为原料经过甲基化或采用boc保护基或cbz 保护基对氮进行保护,然后与十二硫醇进行酯化反应得到甲酸硫酯中间体,甲酸硫酯中间体再经过福山还原得到1-甲基吡咯烷-2-甲醛或boc保护的或 cbz保护的甲醛中间体,boc保护的或cbz保护的甲醛中间体再脱去相应的保护基并甲基化得到1-甲基吡咯烷-2-甲醛,无需将脯氨酸反应成醇再将醇进行氧化得到醛,操作简单,收率高。无需特殊催化剂,价格成本低,整个过程方便操作,最终得到的产品收率高,纯度高。
附图说明
[0032]
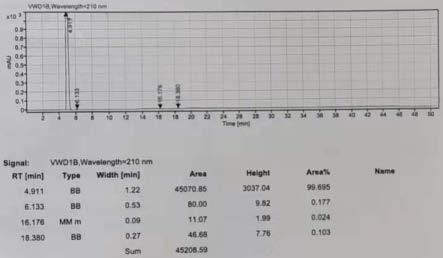
图1为本发明制备的产品的hplc图谱。
具体实施方式
[0033]
下面通过具体实施方式对本发明作进一步详细的说明:
[0034]
本发明的综合反应式为:
[0035][0036]
实施例1
[0037]
中间体z1的合成
[0038]
d-脯氨酸150g,甲醇600ml,多聚甲醛45g,10%pd/c2g,投入3000ml 高压釜中,通入氢气,50℃,反应16个小时左右,采用薄层色谱法监控原料反应完全,过滤,滤液减压浓缩至干,得到白色固体z1 160g,收率94.9%。
[0039]
实施例2
[0040]
中间体z6的合成
[0041]
250ml三口瓶中加入中间体z1 5g(0.0387mol),缩合剂edci(1-乙基
ꢀ‑
3(3-二甲基丙胺)碳二亚胺)8.9g(0.0464mol),dmap(4-二甲氨基吡啶) 0.2g(0.0016mol),加入80ml二氯甲烷,氮气置换后,氮气保护下,降温至0-5℃。滴加预先配置好的十二硫醇二氯甲烷溶液(其中十二硫醇9.4g (0.0464mol)二氯甲烷10ml),保温反应1-1.5小时,加入20ml水洗涤,有机层用50ml饱和食盐水洗涤,5g无水硫酸镁干燥,溶剂旋干,得到黄色油状物z6 12.1g,收率>100%。
[0042]
实施例3
[0043]
中间体z2的合成
[0044]
250ml三口瓶中投入黄色油状物z6 12.1g(0.039mol)、1gpd/c(10%)、 120ml丙
酮,氮气置换后,氮气保护下,降温至0-5℃。滴加13.5g(0.116mol) 三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入50ml二氯甲烷提取,40ml饱和碳酸钠溶液洗涤后,无水硫酸镁干燥后,旋干,得3.8g黄色油状物(z2),收率86.1%。
[0045]
实施例4
[0046]
中间体z5的合成
[0047]
500ml三口瓶中投入d-脯氨酸20g(0.174mol)加入300ml二氯甲烷,室温搅拌下加入19.3g三乙胺(0.19mol),搅拌后,加入二碳酸二叔丁酯41.7g (0.19mol),保温25-30℃反应5-6小时,降温至0℃,用3n盐酸调节ph 值1-2,有机层用饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩至干,得无色油状物,加入石油醚打浆,析出白色固体,过滤,干燥得35.9g白色固体(z5),收率95.8%。
[0048]
实施例5
[0049]
中间体z7的合成
[0050]
250ml三口瓶中加入中间体z5 10g(0.0465mol)、缩合剂edci(1-乙基
ꢀ‑
3(3-二甲基丙胺)碳二亚胺)10.7g(0.0558mol)、dmap(4-二甲氨基吡啶) 0.23g(0.0016mol),加入100ml二氯甲烷,氮气置换后,氮气保护下,降温至0-5℃。滴加预先配置好的十二硫醇二氯甲烷溶液(其中十二硫醇11.3g (0.0558mol)二氯甲烷15ml),保温反应1-1.5小时,加入30ml水洗涤,有机层用60ml饱和食盐水洗涤,5g无水硫酸镁干燥,溶剂旋干,得到黄色油状物z7 18.2g,收率100%。
[0051]
实施例6
[0052]
中间体z8的合成
[0053]
250ml三口瓶中投入黄色油状物z7 18.2g(0.0465mol)、1gpd/c(10%)、 120ml丙酮,氮气置换后,氮气保护下,降温至0-5℃。滴加16.1g(0.14mol) 三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入50ml二氯甲烷提取,40ml饱和碳酸钠溶液洗涤后,无水硫酸镁干燥后,旋干,得8.1g(0.0405mol)黄色油状物(z8),收率87%。
[0054]
实施例7
[0055]
中间体z9的合成
[0056]
500ml三口瓶中投入d-脯氨酸50g(0.434mol)、200ml水,搅拌溶清,加入115g(1.08mol)碳酸钠,搅拌30分钟,降温至0℃左右,滴加预先配置的氯甲酸苄酯甲苯溶液(其中氯甲酸苄酯89g(0.52mol)甲苯90ml),滴加完毕,保温(0℃左右)反应30分钟,升温至20-25℃反应15小时,反应完毕,加入200ml水,过滤,滤液分层,水层用100ml甲苯提取,合并水层,用3n盐酸调ph值至1-2,用200ml乙酸乙酯萃取两次,合并有机相,无水硫酸镁干燥,过滤,减压浓缩至干,得到105g油状物(z9),收率96.7%。
[0057]
实施例8
[0058]
中间体z10的合成
[0059]
250ml三口瓶中加入中间体z9 10g(0.04mol)、缩合剂edci(1-乙基-3(3
‑ꢀ
二甲基丙胺)碳二亚胺)9.2g(0.048mol)、dmap(4-二甲氨基吡啶)0.17g (0.0014mol),加入100ml二氯甲烷,氮气置换后,氮气保护下,降温至0-5℃。滴加预先配置好的十二硫醇二氯甲烷溶液(其中十二硫醇9.7g(0.048mol) 二氯甲烷15ml),保温反应1-1.5小时,加入30ml水洗涤,
有机层用60ml 饱和食盐水洗涤,5g无水硫酸镁干燥,溶剂旋干,得到黄色油状物z10 15.1g,收率87%。
[0060]
实施例9
[0061]
中间体z11的合成
[0062]
250ml三口瓶中投入黄色油状物z10 15.1g(0.0348mol)、1gpd/c(10%)、120ml丙酮,氮气置换后,氮气保护下,降温至0-5℃。滴加12.1g(0.104mol) 三乙基硅烷,滴加完毕,保温反应8-9小时,反应完毕,过滤,滤液减压浓缩至干,加入50ml二氯甲烷提取,40ml饱和碳酸钠溶液洗涤后,无水硫酸镁干燥后,旋干,得7.3g(0.0311mol)黄色油状物(z11),收率89.5%。
[0063]
实施例10
[0064]
中间体z11到中间体z2的合成
[0065]
2000ml高压釜中投入15g中间体z11(0.064mol)、甲醇200ml、多聚甲醛2.3g(0.0736mol)、0.1gpd/c(10%),通入氢气,50℃,反应16个小时左右,监控原料反应完全,过滤,滤液减压浓缩至干,得到油状物6.12g,收率84.5%。
[0066]
实施例11
[0067]
中间体z8到中间体z2的合成
[0068]
100ml单口瓶中投入中间体z8 10g(0.05mol),加入30ml二氯甲烷,室温搅拌下滴加17.1g三氟乙酸(0.15mol),室温反应1-1.5小时,反应完毕,将反应液浓缩至干,加入30ml二氯甲烷,加入15.9g(0.15mol)碳酸钠,搅拌下,滴加入碘甲烷14g(0.1mol),室温下搅拌反应2-3小时,过滤,减压浓缩至干,得到5.4g油状物(z2),收率95.4%。
[0069]
实施例12
[0070]
中间体z3的合成
[0071]
250ml三口瓶中投入中间体z2 10g(0.088mol),二氯甲烷80ml,10-15℃下,滴加入预先配置好的乙氧甲酰基亚甲基三苯基膦的二氯甲烷溶液(其中乙氧甲酰基亚甲基三苯基膦30.6g(0.088mol)二氯甲烷30ml),保温反应 16~18小时,反应完毕后,加入50ml水,用6n盐酸调节ph值至2-3,分区有机层,水层加入100ml二氯甲烷,用4n氢氧化钠调ph值至8-9,取有机层减压浓缩至干,得到酒红色油状物(z3)15.0g,收率93.2%。
[0072]
实施例13
[0073]
中间体z4的合成
[0074]
取中间体(z3)30g(0.164mol),加入4n氢氧化钠溶液82ml,40℃反应 2-3小时,反应完毕,将反应液浓缩至干,用6n盐酸溶液调节ph值至6-7,加入乙酸乙酯萃取,萃取完毕,用6n盐酸调ph值至2-3,将反应液旋干,得到白色固体,异丙醇重结晶,得到产品白色固体z4 24.3g,收率95.6%,纯度99.58%。
[0075]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1