一种来源于威尼斯镰刀菌的启动子及可视化基因敲除筛选方法
1.本发明涉及基因工程领域,具体涉及来源于威尼斯镰刀菌的启动子及可视化基因敲除筛选系统方法。
背景技术:
2.威尼斯镰刀菌是从3000多株真菌中筛选到的可用于发酵生产菌丝蛋白的工业菌株。该菌株发酵产生的菌丝蛋白与单细胞蛋白相比更加味美,具有类似肉质的组织结构,并且能够提供很好的营养平衡,包括脂肪含量低,氨基酸种类齐全,富含微量元素、维生素及利于人胃肠蠕动的可食性粗纤维。此外,威尼斯镰刀菌菌丝蛋白还具有很好的安全性,已在全球的18个国家获得食品原料上市许可。因此,具有部分或全部替代动物性和植物性蛋白质食品的潜力。
3.基因工程是获得优良性状菌株的最有效方法之一。当前在威尼斯镰刀菌中已有相应遗传转化体系的报道,但是由于同源重组率较低(royer, j.c.; christianson, l.m.; yoder, w.t.; gambetta, g.a.; klotz, a.v.; morris, c.l.; brody, h.; otani, s. deletion of the trichodiene synthase gene of fusarium venenatum: two systems for repeated gene deletions. fungal genet. biol.1999,28, 68-78.),导致在原代转化板上筛选靶基因敲除转化体时往往费事费力。
4.因此,开发一套简单的可视化基因敲除转化子筛选系统有望从低同源重组率菌株中方便、快速、高效的筛选到敲除转化子。
技术实现要素:
5.针对现有技术的需求,本发明的目的在于提供来源于威尼斯镰刀菌的高效内源强启动子和适用于威尼斯镰刀菌可视化基因敲除转化子的筛选方法。
6.本发明公开了一种来源于威尼斯镰刀菌的高效内源强启动子,其核苷酸序列如seq id no.1所示。该启动子具体来源于威尼斯镰刀菌tb01,分类命名为镰刀菌fusarium venenatum,于2020年10月12日保藏于中国微生物菌种保藏管理委员会普通微生物中心(cgmcc)(保藏地址为北京市朝阳区北辰西路1号院3号),其保藏编号为cgmcc no.20740。
7.本发明进一步提供包含所述的启动子的重组载体、表达盒。
8.本发明进一步提供一种适于威尼斯镰刀菌的可视化基因敲除的核酸,其特征在于,其包括顺序为启动子元件-荧光蛋白基因-左臂序列-抗性标记基因-右臂序列的核酸片段,其中所述启动子元件是在威尼斯镰刀菌中可高效启动表达的启动子,且与后续的荧光蛋白基因可操作的连接,所述左臂和右臂序列是待敲除基因区域的左臂和右臂序列。优选地,所述启动子元件的核苷酸序列如seq id no:1所示;所述荧光蛋白基因是gfp基因;所述抗性标记基因是neo抗性标记基因。
9.进一步优选地,其是pk2载体为骨架的载体。
10.本发明也提供所述核酸的构建方法,其包括如下步骤:(1)将荧光蛋白基因与所述启动子元件可操作连接后,构建至骨架载体中,获第一重组载体;(2)分别扩增待敲除基因区域的左臂和右臂序列,并将其无缝连接至含有抗性基因的骨架载体的抗性基因元件的两侧,获得第二重组载体;(3)扩增(2)中构建的第二重组载体中的“左臂序列-抗性基因元件-右臂序列”片段,通过同源重组连接到(1)中的第一重组载体上,得到第三重组载体,其元件顺序为启动子元件-荧光蛋白基因-左臂序列-抗性基因-右臂序列;(4)扩增(3)中的启动子元件-荧光蛋白基因-左臂序列-抗性基因-右臂序列的片段即得。
11.本发明尤其提供一种适于威尼斯镰刀菌的可视化基因敲除筛选方法,包括如下步骤:(1)将上述的适于威尼斯镰刀菌的可视化基因敲除的核酸导入到威尼斯镰刀菌原生质体中,优选通过通过peg介导的原生质体转化法;(2)用荧光激发器观察原代转化板,筛选出无荧光的目标基因敲除的转化子。这是因为如果发生了随机插入的同源重组,则菌落呈现荧光;如果实现了目标基因的同源敲除,则菌落不发出荧光。优选地,采用手持荧光激发器进行观察。
12.在具体实施方式中,所述步骤(1)中威尼斯镰刀菌原生质体采用如下方法制备:收集威尼斯镰刀菌孢子,并用无菌0.7 m氯化钠洗涤后,酶解2小时,所述酶解采用的酶裂解液为:20mg崩溃酶+40mg蜗牛酶溶于10ml 0.7 m氯化钠,并过滤除菌。
13.在一个具体实施方式中,所述威尼斯镰刀菌为威尼斯镰刀菌tb01。
14.任选地,还包括通过pcr进一步验证无荧光的菌落,确定是否为目的基因敲除的转化子。
15.在一个具体的实施例中,本发明提供一种可视化基因敲除筛选方法,包括如下步骤:(1)骨架载体pk2-pgpda::gfp的制备;(2)分别扩增待敲除基因区域的左臂和右臂序列,并将其无缝连接至载体pk2-neo的抗性基因表达盒两侧,获得载体pk2-左臂序列-neo-右臂序列;(3)从所述载体pk2-左臂序列-neo-右臂序列中扩增出“左臂序列-neo-右臂序列”片段,通过同源重组连接到步骤(1)中的骨架载体pk2-pgpda::gfp上,得到载体pk2-pgpda::gfp-左臂序列-neo-右臂序列;(4)利用引物对ppklb1/2从载体pk2-pgpda::gfp-左臂序列-neo-右臂序列中扩出“pgpda::gfp-左臂序列-neo-右臂”片段,并通过peg介导的原生质体转化法导入到制备的威尼斯镰刀菌tb01原生质体中;(5)手持荧光激发器观察菌落有无荧光,其中,所述“pgpda::gfp-左臂序列-neo-右臂”片段随机插入到威尼斯镰刀菌tb01的基因组上,则菌落呈现荧光;如果所述“pgpda::gfp-左臂序列-neo-右臂”片段实现目标基因的同源敲除,则菌落不发出荧光。
16.优选的,步骤(1)中骨架载体pk2-pgpda::gfp的制备方法为:以引物对pgfp-t1/2从载体pk2-ptrpc-gfp-ttrpc中扩出gfp-ttrpc序列,以引物对pgpda1/2从威尼斯镰刀菌
tb01的dna基因组中扩出内源启动子pgpda,通过同源重组酶将其连接到抗性骨架载体pk2-neo上获得,其中,所述引物对pgfp-t1/2的上游序列如seq id no.6所示,所述引物对引物对pgfp-t1/2的下游序列如seq id no.7所示,所述引物对pgpda1/2的上游序列如seq id no.4所示,所述引物对引物对pgpda1/2的下游序列如seq id no.5所示,所述启动子pgpda的序列如seq id no:1所示;优选的,所述步骤(4)中威尼斯镰刀菌tb01原生质体采用如下方法制备:收集威尼斯镰刀菌tb01孢子,并用无菌0.7 m氯化钠洗涤后,酶解2小时,所述酶解采用的酶裂解液为:20mg崩溃酶+40mg蜗牛酶溶于10ml 0.7 m氯化钠,并过滤除菌。
17.在一个更具体实施方式中,其包括如下步骤:1)威尼斯镰刀菌tb01高效内源启动子调控gfp基因表达载体的构建及原生质体转化;分别以引物对pgpda1/2(seq id no.4和seq id no.5),pgla1/2(seq id no.10和seq id no.11),ptef1/2(seq id no.12和seq id no.13)从威尼斯镰刀菌tb01的dna基因组中扩出内源启动子pgpda(fvrres_09878,seq id no.1)、pgla(fvrres_01380,seq id no.2)、ptef(fvrres_13282,seq id no.3)的序列,以引物对pgfp-t1/2从载体pk2-ptrpc-gfp-ttrpc-neo(tong, s.;li, m.l.; keyhani, n.o.; liu, y.; yuan, m.; lin, d.m.; jin. d.; li, x.b.; pei, y.; fan, y.h. (2020) characterization of a fungal competition factor: production of a conidial cell-wall associated antifungal peptide. plos pathog.2020,16,e1008518.)中扩出gfp-ttrpc序列。随后通过同源重组酶将其连接到抗性骨架载体pk2-neo上获得相应内源启动子调控gfp基因表达载体。将其进行大肠杆菌dh5α转化后挑选单菌落进行pcr验证,获得阳性转化子。随后通过设计在骨架载体两端的引物对ppklb1/2从pk2-promoter::gfp-neo扩出片段pgpda::gfp-neo,通过peg介导的原生质体转化法导入到转化威尼斯镰刀菌tb01中后,利用手持荧光激发器对各菌株荧光进行荧光观察与比较。
18.2)融合gfp表达盒和靶标基因敲除盒片段的构建及转化分别以引物对几左臂1/2和几右臂1/2从威尼斯镰刀菌tb01的dna基因组中扩出几丁质合成酶基因敲除区域的左臂和右臂序列,通过无缝克隆依次连接到载体pk2-neo(tong, s.;li, m.l.; keyhani, n.o.; liu, y.; yuan, m.; lin, d.m.; jin. d.; li, x.b.; pei, y.; fan, y.h. (2020) characterization of a fungal competition factor: production of a conidial cell-wall associated antifungal peptide. plos pathog.2020,16,e1008518.)的抗性基因表达盒两侧;随后以引物对几敲除盒1/2从构建好的pk2-chs
upstream-neo-chs
downstream
载体中扩出chs
upstream-neo-chs
downstream
片段,并通过同源重组酶(us everbright)将其连接到骨架载体pk2-pgpda::gfp上得到pk2-pgpda::gfp-chs
upstream-neo-chs
downstream
,大肠杆菌dh5α转化后挑选单菌落进行pmei和xbai双酶切验证,挑选正确转化子。随后通过设计在骨架载体两端的引物对ppklb1/2从pk2-pgpda::gfp-chs
upstream-neo-chs
downstream
扩出片段pgpda::gfp-chs
upstream-neo-chs
downstream
,通过peg介导的原生质体转化法导入到转化威尼斯镰刀菌tb01中。如果片段随机插入到威尼斯镰刀菌tb01的基因组上,则菌落呈现荧光;如果片段实现目标基因的同源敲除,则不发出荧光。因此,可以根据手持荧光激发器下菌落有无荧光进行初步筛选。
19.3)可视化基因敲除转化子筛选系统的筛选效率评估。
20.利用手持荧光激发器luyor-3415gr照射原代转化板,筛选出无荧光的候选基因敲除转化子;随后利用菌丝裂解液和中和液对其制备简易模板,并设计可以区分假阳性(扩增条带大小1kb左右)、敲除转化子(扩增条带大小2kb左右)和随机插入(同时包含1kb和2kb左右的条带)的引物,从无荧光转化子中进一步确认阳性敲除转化子。
21.本发明有益效果在于,只需要用手持激发器对原代转化板上照射几秒,就可以轻松排除占绝大多数比例的随机插入转化子(有荧光),随后通过pcr专一性对没有荧光的转化子进行验证,显著提高敲除转化子的筛选效率。本发明提供的筛选系统实现了从低同源重组率菌株中方便、快速、高效的筛选到敲除转化子。
附图说明
22.图1 为内源启动子调控gfp基因表达载体的pcr验证结果图2为不同启动子调控gfp基因表达菌株的菌落荧光观察图3 为pk2-pgpda::gfp-chsupstream-neo-chsdownstream载体的pmei和xbai双酶切验证。其中1-6为提取的大肠杆菌转化子质粒。
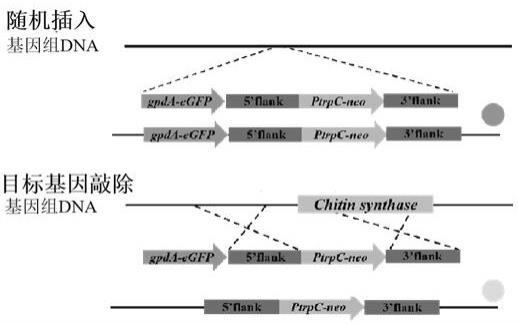
23.图4为可视化筛选系统的工作原理示意图。其中,包含gfp表达盒和几丁质合成酶基因敲除盒的片段如果随机插入到威尼斯镰刀菌tb01的基因组上,则菌落呈现荧光;如果片段实现目标基因的同源敲除,则不发出荧光。
24.图5为可视化基因敲除转化子筛选系统的筛选效率检测。其中a为可视化筛选载体转入威尼斯镰刀菌后荧光初筛的部分结果。b为无荧光候选chs基因敲除转化子的pcr验证结果。菌株wt为野生型威尼斯镰刀菌tb01菌株作为对照,转化为经过荧光初筛的无荧光转化子。
具体实施方式
25.以下实施例进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改或替换,均属于本发明的范围。
26.若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
27.实施例1高效威尼斯镰刀菌tb01内源启动子调控gfp基因表达载体的构建通过调研和分析,参照其他物种中启动子的情况,研究和探索威尼斯镰刀菌tb01的内源启动子pgpda、pgla、ptef,以筛选出内源性强的启动子。具体过程如下:分别以引物对pgpda1/2,pgla1/2,ptef1/2从威尼斯镰刀菌tb01的dna基因组中扩出内源启动子pgpda(fvrres_09878)、pgla(fvrres_01380)、ptef(fvrres_13282)的序列,以引物对pgfp-t1/2从载体pk2-ptrpc-gfp-ttrpc中扩出gfp-ttrpc序列。随后通过同源重组酶将其连接到抗性骨架载体pk2-neo上获得相应内源启动子调控gfp基因表达载体。将其进行大肠杆菌dh5α转化后挑选单菌落进行pcr验证(以gfp片段未检测对象,引物对为gfp1/2),获得阳性转化子。涉及的扩增引物序列参见表1。
28.表1:高效内源启动子调控gfp基因表达载体的构建的引物序列
pgpda1tatgaccatgattacgaattcggtgttgatcgtcaaccaagtcc(seqidno.4)
pgpda2caccatggtggcgacgaatttttgttaaggaggttctgtttgaggaaagat(seqidno.5)pgla1tatgaccatgattacgaattgaatacttgtttcagcgttacaatttccttttga(seqidno.10)pgla2caccatggtggcgacgaattttttattttttttttgctttttctaaatttgattgtatatcctaaggt(seqidno.11)ptef1tatgaccatgattacgaattccacccgtggggagctgtat(seqidno.12)ptef2caccatggtggcgacgaattattgtttaactggatcagtcaagtggtagatag(seqidno.13)pgfp-t1gtcgccaccatggtgagcaa(seqidno.6)pgfp-t2catcttctgtcgacactagtgcctctaaacaagtgtacctg(seqidno.7)gfp1caagctgaccctgaagttca(seqidno.14)gfp2ccttaggacttgtacagctc(seqidno.15)
片段的扩增以及检测的程序参见表2。
29.表2:高效内源启动子调控gfp基因表达载体的构建的引物序列
pgpda片段98℃10s,55℃15s,72℃50s;35个循环 primestar,takarapgla1片段98℃10s,55℃15s,72℃50s;35个循环primestar,takaraptef1片段98℃10s,55℃15s,72℃50s;35个循环primestar,takarapgfp-t片段98℃10s,55℃15s,72℃50s;35个循环primestar,takara转化子验证98℃10s,55℃15s,72℃20s;35个循环primestar,takara同源重组50℃30min
ꢀꢀꢀꢀꢀ
minervasuperfusioncloningkit,useverbright
pcr检测的电泳结果(图1)显示,所选转化子中均扩增出目标条带(对照水中为扩出条带),表明为阳性转化子,后经测序确认后进行-80℃保存。
30.实施例2 不同启动子调控gfp基因表达盒的威尼斯镰刀菌tb01原生质体转化1.培养基yepd:酵母粉3g,蛋白胨10g,葡萄糖20g,定容到1l;溶解酶的缓冲液:0.7 m氯化钠;stc:0.8 m山梨醇;50 mm cacl2,50 mm tris-hcl(ph 8.0);sptc:含40% peg6000的stc;再生培养基:酵母提取物1g,胰蛋白胨1g,蔗糖274g,琼脂糖10g,定容1l;筛选培养基:葡萄糖30g,酵母粉6g,琼脂粉15g,定容到1l。
31.2.引物ppklb1:ggtggcaggatatattgtgg(seq id no.8);ppklb2:ttggttggtcaagtcctggt(seq id no.9)。
32.3.片段扩增程序98℃ 10s, 55℃ 15s, 72℃ 3mins; 35个循环 (primestar,takara)4.实验方法1)转化片段扩增通过设计在骨架载体两端的引物对ppklb1/2从pk2-promoter::gfp-neo扩出片段pgpda::gfp-neo,用于转化威尼斯镰刀菌tb01原生质体。
33.2)原生质体转化a)吐温80制备威尼斯镰刀菌孢悬液,涂于gy固体培养基上,28℃,培养7-10 d产孢。
34.b) 制备孢悬液接于yepd液体培养基中(加玻璃珠),28℃,200rmp培养至萌发菌丝长度为孢子的3-4倍(16h)。
35.c)4℃,13000rpm,15min收集孢子,并用无菌0.7 m氯化钠洗涤1次。
36.d) 酶裂解液制备原生质体(20mg崩溃酶+40mg蜗牛酶溶于10ml 0.7 m氯化钠,并
过滤除菌),30℃ 100 rpm裂解2he) 三层擦镜纸过滤后4℃,7000rpm,10minf) stc洗涤2次,按上述离心后用stc重选原生质体(浓度106),放于冰上备用。
37.g) 取80μl上述原生质体悬浮液,加入20μl sptc,轻轻混匀,加入片段或者线性化载体(300ng/μl)20μl,轻轻混匀后放置冰上30min。
38.h) 加入1ml sptc轻轻混匀,室温放置20min。
39.i) 混合液加到40℃左右的再生培养基中,摇匀倒板,28℃培养过夜(12h左右)j) 倒上筛选培养基,28℃培养长出转化子(3-4d)。
40.3) 不同启动子调控gfp基因表达菌株的荧光观察利用手持荧光激发器luyor-3415gr照射步骤2中获得的不同内源启动子调控gfp基因的阳性菌株,并通过滤片进行拍照。拍照结果参见图2,其中pgpda::gfp菌株荧光最强,即内源启动子pgpda具有最强的表达活性。
41.实施例3 构建可视化基因敲除转化子筛选载体分别以引物对几左臂1/2和几右臂1/2从威尼斯镰刀菌tb01的dna基因组中扩出几丁质合成酶基因敲除区域的左臂和右臂序列,通过无缝克隆依次连接到载体pk2-neo的抗性基因表达盒两侧;随后以引物对几敲除盒1/2从构建好的pk2-chs
upstream-neo-chs
downstream
载体中扩出chs
upstream-neo-chs
downstream
片段,并通过同源重组酶(us everbright)将其连接到骨架载体pk2-pgpda::gfp上得到pk2-pgpda::gfp-chs
upstream-neo-chs
downstream
,大肠杆菌dh5α转化后挑选单菌落进行pmei和xbai双酶切验证,挑选正确转化子。
42.表3构建可视化基因敲除转化子筛选载体涉及引物
几左臂1acagctatgaccatgattacgaattctggtcggttgtgtctggttc(seqidno.16)几左臂2caatgtcatcttctgtcgacactagtggtttggttggtgtgggagtg(seqidno.17)几右臂1gtttagaggtaatccttctttctagagctttcggtcattatcctcc(seqidno.18)几右臂2gcttgcatgcctgcaggtcgactctagagagcgagaaccaactgaaca(seqidno.19)几敲除盒1acaggtacacttgtttagaggcactagttggtcggttgtgtctggttc(seqidno.20)几敲除盒2gcttgcatgcctgcaggtcgactctagagagcgagaaccaactgaaca(同几右臂2,seqidno.19)
表4构建可视化基因敲除转化子筛选载体片段扩增及同源重组程序
几丁质左臂98℃10s,55℃15s,72℃1min;35个循环 primestar,takara几丁质右臂98℃10s,55℃15s,72℃1min;35个循环 primestar,takara几丁质敲除盒98℃10s,55℃15s,72℃2min30s;35个循环 primestar,takara同源重组50℃30min minervasuperfusioncloningkit,useverbright
实验结果参见图3,其中酶切结果显示,2-6号转化子切出目标大小条带(~5kb),为阳性转化子,后经测序确认后进行-80℃保存。
43.实施例4可视化基因敲除转化子筛选载体转化威尼斯镰刀菌tb01原生质体通过设计在骨架载体两端的引物对ppklb1/2从pk2-pgpda::gfp-chs
upstream-neo-chs
downstream
扩出片段pgpda::gfp-chs
upstream-neo-chs
downstream
,其中,引物序列为:ppklb1:ggtggcaggatatattgtgg(seq id no.8);ppklb2:ttggttggtcaagtcctggt(seq id no.9),片段扩增程序:98℃ 10s, 55℃ 15s, 72℃ 4mins; 35个循环(primestar,takara)。用于转化威尼斯镰刀菌tb01原生质体。原生质体转化方法同实施例2。可视化基因敲除转化子筛选原理的示意图参见图4。
44.如果片段随机插入到威尼斯镰刀菌tb01的基因组上,则菌落呈现荧光;如果片段实现目标基因(几丁质合成酶基因chs)的同源敲除,则不发出荧光。因此,可以根据手持荧光激发器下菌落有无荧光进行初步筛选。
45.实施例5可视化基因敲除转化子筛选系统的筛选效率检测利用手持荧光激发器luyor-3415gr照射原代转化板,标记出无荧光的候选基因敲除转化子;随后利用菌丝裂解液和中和液对其制备简易模板,并设计可以区分假阳性(扩增条带大小1kb左右)、敲除转化子(扩增条带大小2kb左右)和随机插入的引物(其中,验证引物1:cactcccacaccaaccaaac(seq id no.21);验证引物2:ggtcaggaggataatgaccg(seq id no.22),片段扩增程序98℃ 10s, 55℃ 15s, 72℃ 50s; 35个循环 (primestar,takara)),从无荧光转化子中进一步确认阳性敲除转化子。其中,菌丝裂解液:取1.2 gnaoh用去离子水溶解后定容至100 ml,菌丝中和液:10 ml 1m tris-hcl(ph8.0),40 ml 0.3 m hcl,用去离子水定容至800 ml。
46.实验结果参见图5,其中,通过荧光初步筛选,从72个转化子中筛选到19个无荧光的菌落(图5中a)。随后pcr进一步验证发现,19个无荧光菌落中有13个为敲除转化子(图5中b)。上述结果表明,相比于直接pcr筛选敲除转化子(13/72),可视化筛选系统的筛选效率(13/19)提高了4倍左右。
47.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1