多羟基吡咯烷类化合物及其制备方法和应用
1.本发明涉及糖苷酶抑制剂领域,具体涉及一种多羟基吡咯烷类化合物及其制备方法和应用。
背景技术:
2.亚氨基糖广泛的分布在植物的体内,天然亚氨基糖根据其骨架结构可以分为多羟基吡咯烷、多羟基哌啶、多羟基吲哚里西啶、多羟基吡咯里西啶和多羟基去甲基托品烷。它们对不同的糖苷酶表现出良好的抑制活性,如小构树碱是从小构树中分离出的一系列天然产物,它对β-葡萄糖苷酶和β-半乳糖苷酶有很好的抑制活性,而其对映体则表现出良好的α-葡萄糖苷酶抑制活性[(a)m.shibano,s.kitagawa,g.kusano,chem.pharm.bull.1997,45,505-508;(b)m.shibano,s.kitagawa,s.nakamura,n.akazawa,g.kusano,chem.pharm.bull.1997,45,700-705;(c)m.shibano,s.nakamura,n.akazawa,g.kusano,chem.pharm.bull.1998,46,1048-1050;(d)m.shibano,s.nakamura,m.kubori,k.minoura,g.kusano,chem.pharm.bull.1998,46,1416-1420;(e)m.shibano,s.nakamura,n.motoya,g.kusano,chem.pharm.bull.1999,47,472-476;(f)m.shibano,d.tsukamoto,g.kusano,chem.pharm.bull.1999,47,907-908;(g)m.shibano,d.tsukamoto,g.kusano,heterocycles 2002,57,1539-1553.]。这样的特性使其在抗病毒、治疗溶酶体贮积症、高血氏病以及糖尿病等方面具有潜在的药用价值。因此,对多羟基吡咯烷化合物的深入研究意义重大。
技术实现要素:
[0003]
本发明的目的是为了提供一种多羟基吡咯烷类化合物及其制备方法和应用,该多羟基吡咯烷类化合物具有较高的糖苷酶的抑制活性,尤其是对β-葡萄糖苷酶表现出优异的抑制活性,能够用于预防和治疗多种相关疾病。
[0004]
为了实现上述目的,本发明第一方面提供一种多羟基吡咯烷类化合物,所述多羟基吡咯烷类化合物的结构式如式(1)所示:
[0005][0006]
其中,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c10的环烷基、c1-c10的烷氧基以及c1-c10的直链或支链烷基中的一种;
[0007]
n为1-20的整数;
[0008]
*表示手性碳原子。
[0009]
本发明第二方面提供一种多羟基吡咯烷类化合物的制备方法,该方法包括以下步骤:
[0010]
1)将式(2)所示硝酮与具有乙烯基的有机金属试剂在第一亲核加成反应条件下进行第一亲核加成反应,得到式(3)所示羟胺,
[0011][0012]
其中,r6为苄基或者苯环上的氢被羟基、烷氧基、硝基、卤素单一至全取代的苄基;
[0013]
*表示手性碳原子;
[0014]
2)将步骤1)所得式(3)所示羟胺与还原剂在还原反应条件下进行还原反应,并将还原反应所得产物与氨基保护剂在氨基保护反应条件下进行氨基保护反应,得到式(4)所示化合物,
[0015][0016]
其中,r7为来自所述氨基保护剂的残基;
[0017]
优选地,r7为叔丁氧羰基、苄氧羰基、芴氧羰基或乙酰基;
[0018]
3)将步骤2)所得式(4)所示化合物与过氧化物在双键环氧化反应条件下进行双键环氧化反应,得到式(5)所示化合物和式(6)所示化合物的混合物,
[0019][0020]
4)将来自于步骤(3)所得混合物中的式(5)所示化合物或式(6)所示化合物与具有-ch
2-(ch2)
m-r8的有机金属试剂在第二亲核加成反应条件下进行第二亲核加成反应,得到式(7)所示化合物,
[0021][0022]
其中,m为0-18的整数;
[0023]
r8为-ch=ch2或-ch=chx,此处,x为卤素或取代硼烷,取代硼烷中的取代基选自c1-6的烷基、c1-6的烷氧基和氟硼酸钾中的一种;
[0024]
5)将步骤4)所得式(7)所示化合物与式(8)所示化合物在偶联反应条件下进行偶联反应,得到式(9)所示化合物,
[0025]
[0026]
其中,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c10的环烷基、c1-c10的烷氧基以及c1-c10的直链或支链烷基中的一种;
[0027]
6)将式(9)所示化合物与氢源在催化氢化反应条件下进行催化氢化反应,得到式(1)所示化合物,
[0028][0029]
其中,n为0-20的整数,n=m+2。
[0030]
本发明第三方面提供一种糖苷酶抑制剂,其中,所述糖苷酶抑制剂含有本发明第一方面所述的多羟基吡咯烷类化合物或本发明第二方面所述制备方法制备得到的多羟基吡咯烷类化合物。
[0031]
优选地,所述糖苷酶抑制剂用于抑制α-葡萄糖苷酶、β-葡萄糖苷酶、α-半乳糖苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-甘露糖苷酶、α-l-岩藻糖苷酶、α,α-海藻糖酶、α-l-鼠李糖苷酶、淀粉葡糖苷酶和β-葡糖苷酸化酶中的一种或多种;更优选地,所述糖苷酶抑制剂用于抑制β-葡萄糖苷酶;进一步优选地,所述糖苷酶抑制剂用于抑制牛肝来源的β-葡萄糖苷酶。
[0032]
本发明第四方面提供一种本发明第一方面所述的多羟基吡咯烷类化合物或本发明第二方面所述制备方法制备得到的多羟基吡咯烷类化合物制备药物中的应用,所述药物选自下述药物中的至少一种:1)预防和/或治疗糖尿病的药物;2)预防和/或治疗高雪氏病的药物;3)预防和/或治疗溶酶体贮积症的药物;4)预防和/或治疗肿瘤的药物;5)抗病毒药物。
[0033]
本发明提供的多羟基吡咯烷类化合物具有良好的糖苷酶、尤其是β-葡萄糖苷酶的抑制活性,同时对于β-葡萄糖苷酶还呈现良好的选择性。因此具有预防和治疗糖尿病、高雪氏病、溶酶体贮积症以及肿瘤等多种疾病的潜力,还可以应用于抗病毒药物。
[0034]
另外,本发明提供的多羟基吡咯烷类化合物对于牛肝来源的β-葡萄糖苷酶表现出非常优异的抑制活性。
[0035]
通过本发明提供的测试例可以看出,本发明中的式(1-1)所示化合物和式(1-3)所示化合物显示出了非常显著的对于牛肝来源的β-葡萄糖苷酶抑制活性,与其它化合物相比抑制活性提高了十倍以上,甚至可以高达近千倍。与此同时,式(1-1)所示化合物和式(1-3)所示化合物还表现出明显更低的大鼠小肠麦芽糖酶来源的α-葡萄糖苷酶抑制活性,由此表明式(1-1)所示化合物和式(1-3)所示化合物作为药物组分摄入后对肠道的影响显著降低,从而会大大降低产生腹泻等副作用的几率和程度。十分适合作为药物组分应用。
[0036]
另外,本发明提供的多羟基吡咯烷类化合物的制备方法简单易行,收率和纯度均较高,非常适用于工业生产。
具体实施方式
[0037]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各
个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0038]
本发明第一方面提供一种多羟基吡咯烷类化合物,其中,所述多羟基吡咯烷类化合物的结构式如式(1)所示:
[0039][0040]
式(1)中,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c10的环烷基、c1-c10的烷氧基以及c1-c10的直链或支链烷基中的一种;
[0041]
n为1-20的整数;
[0042]
*表示手性碳原子。
[0043]
本发明中,卤素例如可为氟、氯、溴或碘。
[0044]
本发明中,c3-c10的环烷基例如可以为环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、环癸基等。
[0045]
本发明中,c1-c10的烷氧基例如可以为甲氧基、乙氧基、丙氧基、丁氧基、戊氧基、己氧基、庚氧基、辛氧基、壬氧基、癸氧基等。
[0046]
本发明中,c1-c10的直链或支链烷基例如可以为甲基、乙基、丙基、异丙基、异丁基、仲丁基、叔丁基、异戊基、异己基、异庚基、2-甲基己基、2-乙基己基、1-甲基庚基、2-甲基庚基、异辛基、异壬基或3,5,5-三甲基己基等。
[0047]
根据本发明的第一方面,式(1)中,优选地,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c8的环烷基、c1-c8的烷氧基以及c1-c8的直链或支链烷基中的一种;更优选地,r1、r2、r3、r4和r5各自独立地选自氢、氟、氯、溴、氨基、环丙基、环丁基、环戊基、环己基以及c1-c6的直链或支链烷基中的一种;进一步优选地,r1、r2、r3、r4和r5各自独立地选自氢、氟、氯、甲基和叔丁基中的一种。
[0048]
另外,优选地,r1、r2、r3、r4和r5不全为氢。
[0049]
本发明中,所述n例如可以为0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20,没有特别的限定。
[0050]
优选地,n为1-10的整数;更优选地,n为1-6的整数。
[0051]
根据本发明一个特别优选地实施方式,其中,n为5。
[0052]
本发明中,优选地,*各自独立地为r或s构型;
[0053]
另外,优选地,式ⅰ所示化合物的2、3、4、5位碳的立体构型为2r、3r、4r、5r或2s、3s、4s、5s。
[0054]
本发明中,优选地,式ⅰ所示的化合物的结构为以下结构中的任意一种:
[0055][0056][0057]
其中,f为氟。
[0058]
当式ⅰ所示的化合物为式(1-1)至式(1-20)所示的结构中的任意一种时,其具备较
好的糖苷酶抑制活性,尤其是对β-葡萄糖苷酶表现出良好的抑制活性,并且对于牛肝来源的β-葡萄糖苷酶表现出非常优异的抑制活性。
[0059]
另外,式(1-1)所示化合物和式(1-3)所示化合物显示出了非常显著的对于牛肝来源的β-葡萄糖苷酶抑制活性,与其它化合物相比抑制活性显著提高。同时式(1-1)所示化合物和式(1-3)所示化合物还表现出明显较低的大鼠小肠麦芽糖酶来源的α-葡萄糖苷酶抑制活性,表明式(1-1)所示化合物和式(1-3)所示化合物作为药物组分摄入后对肠道的影响显著降低,从而会大大降低产生腹泻等副作用的几率和程度。十分适合作为药物组分应用。
[0060]
本发明第二方面提供本发明第一方面所述的多羟基吡咯烷类化合物的制备方法,该方法包括以下步骤:
[0061]
1)将式(2)所示硝酮与具有乙烯基的有机金属试剂在第一亲核加成反应条件下进行第一亲核加成反应,得到式(3)所示羟胺,
[0062][0063]
其中,r6为苄基或者苯环上的氢被羟基、烷氧基、硝基、卤素单一至全取代的苄基;
[0064]
*表示手性碳原子;
[0065]
2)将步骤1)所得式(3)所示羟胺与还原剂在还原反应条件下进行还原反应,并将还原反应所得产物与氨基保护剂在氨基保护反应条件下进行氨基保护反应,得到式(4)所示化合物,
[0066][0067]
其中,r7为来自所述氨基保护剂的残基;
[0068]
3)将步骤2)所得式(4)所示化合物与过氧化物在双键环氧化反应条件下进行双键环氧化反应,得到式(5)所示化合物和式(6)所示化合物的混合物,
[0069][0070]
4)将来自于步骤(3)所得混合物中的式(5)所示化合物或式(6)所示化合物与具有-ch
2-(ch2)
m-r8的有机金属试剂在第二亲核加成反应条件下进行第二亲核加成反应,得到式(7)所示化合物,
[0071][0072]
其中,m为0-18的整数;
[0073]
r8为-ch=ch2或-ch=chx,此处,x为卤素或取代硼烷,取代硼烷中的取代基选自c1-6的烷基、c1-6的烷氧基和氟硼酸钾中的一种;
[0074]
5)将步骤4)所得式(7)所示化合物与式(8)所示化合物在偶联反应条件下进行偶联反应,得到式(9)所示化合物,
[0075][0076]
其中,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c10的环烷基、c1-c10的烷氧基以及c1-c10的直链或支链烷基中的一种;
[0077]
6)将式(9)所示化合物与氢源在催化氢化反应条件下进行催化氢化反应,得到式(1)所示化合物,
[0078][0079]
其中,n为0-20的整数,n=m+2。
[0080]
以下,对所述多羟基吡咯烷类化合物的制备方法进行具体地说明。
[0081]
首先,对步骤1)所述第一亲核加成反应进行说明。
[0082]
步骤1)中,将式(2)所示的硝酮具有乙烯基的有机金属试剂进行第一亲核加成反应,得到式(3)所示的羟胺来作为所述多羟基吡咯烷类化合物的骨架结构。
[0083]
本发明中,式(2)所示硝酮可以具有下述式(2-1)和式(2-2)所示的对映体,也就是说,在本发明中,式(2)所示硝酮可以是仅为式(2-1)所示的结构的化合物,也可以是仅为式(2-2)所示结构的化合物,还可以是式(2-1)和式(2-2)所示结构的化合物的混合物。
[0084][0085]
式(2)所示硝酮可以采用本领域常规的方法制备而成,也可以商购获得。以r6为苄基为例,式(2-1)或式(2-2)所示硝酮可以购自百灵威科技有限公司。
[0086]
本发明中,优选地,r6为苄基。
[0087]
根据本发明,步骤1)中,所述第一亲核加成反应在第一溶剂的存在下进行;优选地,所述第一溶剂为无水非质子溶剂;更优选地,所述第一溶剂为醚类溶剂和/或卤代烷基溶剂;进一步优选地,所述第一溶剂为乙醚、四氢呋喃、二氧六环和二氯甲烷中的一种或多种。
[0088]
本发明中,所述第一溶剂的用量可以根据式(2)所示硝酮的量来决定。优选地,相对于1mmol的式(2)所示硝酮,所述第一溶剂的用量为1-6ml,更优选为2-4ml,由此可以促进反应的进行。
[0089]
根据本发明,所述具有乙烯基的有机金属试剂优选为具有乙烯基的有机镁试剂、具有乙烯基的有机锌试剂、具有乙烯基的有机锂试剂、具有乙烯基的有机铜试剂和具有乙烯基的有机硅试剂中的一种或多种;更优选地,所述具有乙烯基的有机金属试剂为乙烯基氯化镁、乙烯基溴化镁、乙烯基锂和乙烯基溴化锌中的一种或多种。
[0090]
在本发明一个特别优选地实施方式中,所述具有乙烯基的有机金属试剂为乙烯基溴化镁,由此可以进一步提高反应效率。
[0091]
本发明中,所述具有乙烯基的有机金属试剂的用量可以根据式(2)所示硝酮的量来决定。例如,式(2)所示硝酮与所述具有乙烯基的有机金属试剂的摩尔比可以为1:1.2-3,优选为1:1.5-2.5。由此可以提高反应效率,保证制备得到的式(3)所示羟胺的收率。
[0092]
根据本发明,所述第一亲核加成反应的条件可以包括:温度为-80℃到20℃,时间为0.2-3h;优选地,所述第一亲核加成反应的条件包括:温度为-10℃到10℃,时间为0.5-2h。通过在上述条件下进行所述第一亲核加成反应,可以促进反应的顺利进行,提高反应效率和收率。
[0093]
另外,本发明中,优选地,所述第一亲核加成反应在惰性气体的保护下进行,所述惰性气体例如可以为氮气、氩气、氦气、氖气等。
[0094]
另外,在步骤1)所述第一亲核加成反应结束后,可以采用本领域的常规方法对反应进行淬灭,例如可以使用饱和nh4cl的水溶液进行淬灭,淬灭后可以采用萃取等方法对淬灭的产物进行精制。例如,萃取可以使用乙酸乙酯,根据需要萃取后可以对有机相进行洗涤、干燥和浓缩。此为本领域的常规操作,此处不再详细说明。
[0095]
接着,对步骤2)所述还原反应和氨基保护反应进行说明。
[0096]
步骤2)中,先将步骤1)所得式(3)所示羟胺与还原剂在还原反应条件下进行还原反应,得到式(3’)所示的胺。
[0097][0098]
其中,所述还原反应在第二溶剂的存在下进行。
[0099]
为了使得还原剂与后述活化剂更好地反应,从而更顺利地进行所述还原反应,优选地,所述第二溶剂为酸性溶剂。
[0100]
更优选地,所述第二溶剂为乙酸、丙酸、丁酸和甲酸中的一种或多种,进一步优选为乙酸。
[0101]
此外,所述第二溶剂的用量可以根据式(3)所示羟胺的量来决定,例如,相对于1mmol式(3)所示羟胺,所述第二溶剂的用量可以为20-80ml。为了更利于所述还原反应的进行,相对于1mmol式(3)所示羟胺,所述第二溶剂的用量优选为30-50ml。
[0102]
根据本发明,所述还原剂可以使用本领域通常用于将羟胺还原成胺的各种还原剂,例如可以使用锌粉、铁粉和氢化铝锂中的一种或多种;优选地,所述还原剂为锌粉。由此可以提高还原反应的速率。
[0103]
此外,为了保证还原反应的充分、顺利进行,所述还原剂的用量也可以根据式(3)所示羟胺的量来决定,例如,式(3)所示羟胺与还原剂的摩尔比可以为1:5-20;优选为1:10-15。
[0104]
另外,本发明中,为了促进反应的进行,可以提前对所述还原剂进行活化,或者在所述还原反应体系中加入活化剂。
[0105]
本发明中,优选地,提前对所述还原剂进行活化。提前对所述还原剂进行活化时,可以采用本领域常规的活化方式进行,没有特别的限定,例如,可以采用盐酸对所述还原剂进行活化。
[0106]
本发明中对于所述活化剂没有特别的限定,可以为本领域通常用于将还原剂活化的各种活化剂,例如可以为cu(oac)2、agoac、cuso4和cucl2中的一种或多种;优选地,所述活化剂为cu(oac)2和/或agoac。
[0107]
根据本发明,为了对所述还原剂进行充分地活化,所述活化剂的用量可以根据所述还原剂的用量来决定,例如,所述活化剂与所述还原剂的摩尔比可以为1:80-200;优选为1:100-200。
[0108]
本发明中,所述还原反应的条件可以包括:温度为10-30℃,时间为2-12h;优选地,所述还原反应的条件包括:温度为20-30℃,时间为2-6h。
[0109]
根据本发明,在所述还原反应结束后,可以采用本领域通常用于精制的各种方法对所述还原反应得到的产物进行精制。例如,可以将所述还原反应产物进行浓缩后,使用有机溶剂进行分散,并对其进行洗涤,洗涤可以使用碱性溶液进行,例如可以使用饱和碳酸氢钠水溶液,在使用碱性溶液进行洗涤后,根据需要还可以使用水进行洗涤,并且,根据需要还可以对洗涤后的水相进行萃取以提高收率,然后可以将有机相进行干燥(例如可以使用无水硫酸钠进行干燥)、浓缩得到式(3’)所示结构的胺的粗产物。
[0110]
在本发明中,可以将所述粗产物直接用于下一步反应。
[0111]
接着,步骤2)中,将还原反应所得产物,即(3’)所示结构的胺氨基保护剂在氨基保护反应条件下进行氨基保护反应,得到式(4)所示化合物。
[0112]
本发明中,步骤2)所述反应会产生酸,因此,为了中和反应中产生的酸,促进反应的进行,优选地,所述氨基保护反应在第一碱的存在下进行。
[0113]
所述第一碱可以为本领域常用的各种无机碱或有机碱,优选为无机碱。
[0114]
作为所述第一碱的无机碱例如可以为碳酸氢钠、氢氧化钠、碳酸氢钾和氢氧化钾中的一种或多种;更优选地,所述第一碱为碳酸氢钠。
[0115]
本发明中,所述第一碱的用量可以根据所述还原反应产物的量来决定,例如,所述还原反应所得产物与所述第一碱的摩尔比可以为1:1.0-3.0;优选为1:1.5-2.0。
[0116]
另外,本发明中,所述氨基保护反应在第三溶剂的存在下进行。
[0117]
所述第三有机溶剂可以采用本领域常见的各种有机溶剂,没有特别的限制。
[0118]
但是,当使用所述第一碱,且所述第一碱为无机碱时,为了增加无机碱的溶解度,优选地,所述第三溶剂为有机溶剂与水的混合溶剂;例如,所述第三溶剂可以为乙醚、四氢呋喃、二氧六环和二氯甲烷中的一种或多种与水的混合溶剂;优选地,所述第三溶剂为四氢呋喃和水的混合溶剂。
[0119]
此时,优选地,所述第三溶剂中,有机溶剂和水的比例可以为1-10:1,优选为2-8:1。
[0120]
本发明中,所述第三溶剂的用量可以根据所述还原反应所得产物的量来决定。例如,相对于1mmol所述还原反应所得产物,所述第三溶剂的用量为5-20ml;优选为8-12ml。
[0121]
根据本发明,所述氨基保护剂可以为氯甲酸苄酯、芴甲氧羰酰氯、氯甲酸烯丙酯和碳酸二叔丁酯中的一种或多种;优选地,所述氨基保护剂为氯甲酸苄酯。
[0122]
优选地,式(4)所示化合物中氨基保护基r7为苄氧羰基。
[0123]
本发明中,所述还原反应所得产物与所述氨基保护剂的摩尔比为1:1.0-3.0;优选为1:1.5-2.0。
[0124]
另外,本发明所述氨基保护反应的条件没有特别的限定,例如,所述氨基保护反应的条件可以包括:温度为0-40℃,时间为0.2-5h;为了进一步促进所述氨基保护反应的进行,优选地,所述氨基保护反应的条件可以包括:温度为0-30℃,时间为0.5-2h。
[0125]
以下,对步骤3)所述双键环氧化反应进行具体描述。
[0126]
步骤3)中,将步骤2)所得式(4)所示化合物与过氧化物在双键环氧化反应条件下进行双键环氧化反应。
[0127]
本发明步骤3)中,所述双键环氧化反应在第四溶剂的存在下进行。
[0128]
所述第四溶剂优选为有机溶剂,例如可以为二氯甲烷、1,2-二氯乙烷、叔丁醇、乙腈、丙腈和丁腈中的一种或多种;优选地,所述第四溶剂为二氯甲烷。
[0129]
根据本发明,所述第四溶剂的量可以根据式(4)所示化合物的量来决定,例如,相对于1mmol式(4)所示化合物,所述第四有机溶剂的用量为5-30ml;优选地为10-20ml,由此可以进一步促进反应的顺利进行。
[0130]
另外,为了中和反应过程中产生的酸性物质,促进反应的顺利进行,提高反应效率和收率,优选地,所述双键环氧化反应在第二碱的存在下进行。
[0131]
此处,所述第二碱例如可以为碳酸氢钠、磷酸氢二钠、碳酸钠、碳酸钾、碳酸氢钾和磷酸氢二钾中的一种或多种;优选为碳酸氢钠。
[0132]
本发明中,所述第二碱的用量也可以根据式(4)所示化合物的量决定,例如,式(4)所示化合物与所述第二碱的摩尔比可以为1:1-5;优选为1:2-3。
[0133]
根据本发明,步骤3)中所述过氧化物为间氯过氧苯甲酸、过氧化氢、过氧乙酸、oxone和过氧叔丁醇中的一种或多种;优选为间氯过氧苯甲酸。由此更有利于所述双键环氧化反应的进行,提高收率。
[0134]
此外,所述过氧化物的用量可以根据式(4)所示化合物的用量来决定,例如,式(4)所示化合物与所述过氧化物的摩尔比可以为1:1-5;优选地,式(4)所示化合物与所述过氧化物的摩尔比为1:2-3。
[0135]
另外,本发明中,所述双键环氧化反应的条件可以包括:温度为5-50℃,时间为1-6天;优选地,温度为10-40℃,时间为2-4天。
[0136]
由此,通过所述双键环氧化反应,得到式(5)所示化合物和式(6)所示化合物的混合物。
[0137]
反应完成后,可以采用本领域常规的分离方法将所述混合物中的式(5)所示化合物和式(6)所示化合物进行分离。根据目标产物选择式(5)所示化合物或式(6)所示化合物进行后续步骤。
[0138]
所述分离方法例如可以包括柱层析、打浆和萃取等,此为本领域的常规操作,本说明书中不再详细描述。
[0139]
接着,对步骤4)所述第二亲核加成反应进行说明。
[0140]
步骤4)中,将来自于步骤(3)所得混合物中的式(5)所示化合物或式(6)所示化合物与具有-ch
2-(ch2)
m-r8的有机金属试剂进行第二亲核加成反应,以得到式(7)所示化合物。
[0141]
本发明中,为了使反应更顺利进行,所述第二亲核加成反应在第五有机溶剂的存在下进行。
[0142]
优选地,所述第五有机溶剂为无水非质子溶剂;更优选地,所述第五溶剂为乙醚、四氢呋喃、二氧六环和二氯甲烷中的一种或多种。
[0143]
另外,相对于1mmol式(5)所示化合物或式(6)所示化合物,所述第五有机溶剂的用量可以为5-20ml,优选为10-15ml。
[0144]
根据本发明,为了促进第二亲核加成反应的进行,优选地,所述第二亲核加成反应在第一催化剂的存在下进行。
[0145]
所述第一催化剂可以为本领域常见的进行亲和加成反应的各种催化剂,例如可以为碘化亚铜、氰化亚铜、氯化亚铜和溴化亚铜中的一种或多种;优选为碘化亚铜。
[0146]
另外,所述第一催化剂的用量可以根据式(5)所示化合物或式(6)所示化合物的量来调整,例如,式(5)所示化合物或式(6)所示化合物与所述第一催化剂的摩尔比为1:0.05-0.4;优选为1:0.08-0.15。由此可以提高所述第二亲核加成反应的效率。
[0147]
本发明中,所述具有-ch
2-(ch2)
m-r8的有机金属试剂例如可以为有机镁试剂、有机锌试剂、有机锂试剂、有机铜试剂和有机硅试剂中的一种或多种。
[0148]
如上所述,其中r8可以为-ch=ch2或-ch=chx,此处,x为卤素或取代硼烷。
[0149]
所述卤素例优选为氯、溴或碘,所述取代硼烷中的取代基例如可以选自c1-6的烷基、c1-6的烷氧基和氟硼酸钾中的一种。
[0150]
本发明中,优选地,所述r8为-ch=ch2。
[0151]
另外,具有-ch
2-(ch2)
m-r8的有机金属试剂中,m为0-18的整数,可以根据目标产物(即多羟基吡咯烷类化合物)来对所述m的取值进行选择。例如,为了制备式(1-1)所示的化合物,具有-ch
2-(ch2)
m-r8的有机金属试剂中的m则应该选择为2。
[0152]
根据本发明,优选地,所述具有-ch
2-(ch2)
m-r8的有机金属试剂为mgbr-ch
2-(ch2)
2-ch=ch2、mgbr-ch
2-(ch2)
3-ch=ch2和znbr-ch
2-(ch2)
2-ch=ch2中的一种或多种。
[0153]
另外,此处所述具有-ch
2-(ch2)
m-r8的有机金属试剂可以采用本领域常规的方法进行制备。以有机镁试剂mgbr-ch
2-(ch2)
2-ch=ch为例,可以采用-ch
2-(ch2)
2-ch=ch2的卤代物,即x-ch
2-(ch2)
2-ch=ch2(x为氯、溴或碘)与镁进行反应制得,本发明就此不再赘述。
[0154]
本发明中,式(5)所示化合物或式(6)所示化合物与所述具有-ch
2-(ch2)
m-r8的有机金属试剂的摩尔比可以为1:1.2-3;优选为1:1.5-2.0。
[0155]
根据本发明,所述第二亲核加成反应的条件可以包括:温度为-10~30℃,时间为0.5-2h;优选地,所述第二亲核加成反应的条件包括:温度为10-25℃,时间为0.5-1h。
[0156]
此外,在步骤4)所述第二亲核加成反应结束后,当然也可以采用本领域的常规方法对反应进行淬灭,例如可以使用饱和nh4cl的水溶液进行淬灭,淬灭后可以采用萃取等方法对淬灭的产物进行精制。例如,萃取可以使用乙酸乙酯,根据需要萃取后可以对有机相进行洗涤、干燥和浓缩。此处不再详细说明。
[0157]
由此,得到式(7)所示化合物。
[0158]
接着,对步骤5)所述偶联反应进行说明。
[0159]
步骤5)中,将步骤4)所得式(7)所示化合物与式(8)所示化合物进行偶联反应,得到式(9)所示化合物。
[0160]
式(8)中,r1、r2、r3、r4和r5与本发明第一方面所述内容一致,可以根据目标产物来对应地进行选择。
[0161]
优选地,r1、r2、r3、r4和r5各自独立地选自氢、卤素、氨基、c3-c8的环烷基、c1-c8的烷氧基以及c1-c8的直链或支链烷基中的一种;更优选地,r1、r2、r3、r4和r5各自独立地选自氢、氟、氯、溴、氨基、环丙基、环丁基、环戊基、环己基以及c1-c6的直链或支链烷基中的一种;进一步优选地,r1、r2、r3、r4和r5各自独立地选自氢、氟、氯、甲基和叔丁基中的一种。
[0162]
另外,优选地,r1、r2、r3、r4和r5不全为氢。
[0163]
根据本发明,步骤5)所述偶联反应在第六溶剂的存在下进行。
[0164]
所述第六溶剂没有特别的限定,可以为本领域常规的各种用于进行偶联反应的溶剂。例如,所示第六溶剂可以为二氯甲烷、1,2-二氯乙烷和甲苯中的一种或多种。优选地,所述第六溶剂为二氯甲烷。
[0165]
所述第六溶剂的量可以根据式(7)所示化合物的量进行调整,例如,相对于1mmol式(7)所示化合物,所述第六溶剂的用量为10-50ml;优选为20-30ml。
[0166]
此外,根据本发明,所述偶联反应还可以在第二催化剂的存在下进行。
[0167]
对于所述第二催化剂,例如可以为本领域常见的用于偶联反应的催化剂,例如,所述第二催化剂可以为钯催化剂、镍催化剂、grubbs催化剂和hoveyda-grubbs催化剂中的一种或多种。
[0168]
本发明的发明人在试验过程中发现,当所述催化剂为hoveyda-grubbs时,可以大幅地提高偶联的产率以及反应速率,从而提高制备的多羟基吡咯烷类化合物的产率,非常适用于本技术所述的偶联反应。
[0169]
因此,本发明中,特别优选地,采用第二代hoveyda-grubbs催化剂作为第二催化剂进行所述偶联反应,该催化剂可以通过商购获得。
[0170]
根据本发明,所述第二催化剂的用量可以根据式(7)所示化合物与式(8)所示化合物的总量来选择,优选地,式(7)所示化合物与式(8)所示化合物的总量与所述第二催化剂的摩尔比为1:0.03-0.08;更优选为1:0.04-0.06。由此可以保证偶联反应的反应速率,提高收率。
[0171]
另外,本发明中,为了提高收率,式(7)所示化合物与式(8)所示化合物的摩尔比可以为1:1.5-15;优选为1:3-10。
[0172]
此外,所述偶联反应优选在惰性气体保护下进行,例如,可以在氮气、氩气、氦气等气体的保护下进行。
[0173]
根据本发明,所述偶联反应的条件可以为本领域的常规条件,例如,所述偶联反应的条件包括:温度为30-50℃,时间为2-24h;优选地,所述偶联反应的条件包括:温度为40-45℃,时间为6-12h。
[0174]
通过上述偶联反应,将式(7)所示化合物与式(8)所示化合物进行偶联,得到式(9)所示化合物。
[0175]
接着,对步骤6)的催化氢化反应进行说明。
[0176]
步骤6)中,将式(9)所示化合物与氢源进行催化氢化反应,得到式(1)所示化合物。
[0177]
根据本发明,步骤6)中,所述催化氢化反应在第七溶剂的存在下进行。
[0178]
所述第七溶剂没有特别的要求,可以为本领域的常规选择,例如可以为甲醇、乙醇、乙酸乙酯、四氢呋喃和水中的一种或多种;优选为甲醇。
[0179]
另外,所述第七溶剂的用量可以根据式(9)所示化合物的量来决定。例如,相对于1mmol的式(9)所示化合物,所述第七有机溶剂的用量可以为10-50ml;优选地,相对于1mmol的式(9)所示化合物,所述第七有机溶剂的用量可以为20-40ml。
[0180]
根据本发明,优选地,所述催化氢化反应在第三催化剂的存在下进行。
[0181]
所述第三催化剂可以采用本领域常规的用于进行催化加氢的催化剂,例如可以为pd/c、氢氧化钯、氧化铂和雷尼镍中的一种或多种,没有特别的限定。优选地,所述第三催化剂为pd/c。
[0182]
此外,所述第三催化剂的用量可以根据式(9)所示化合物的量来选择,例如,式(9)所示化合物与所述第三催化剂的摩尔比可以为1:0.03-0.2;优选为1:0.05-0.1。
[0183]
本发明中,所述氢源没有特别的限定,例如可以为氢气、甲酸铵、甲酸、乙酸铵、硼氢化钠、环己烯和环己二烯中的一种或多种;优选为氢气。
[0184]
所述氢源的用量根据式(9)所示化合物的量来进行选择,例如,式(9)所示化合物与所述氢源的摩尔比可以为1:20-100;优选为1:50-70。
[0185]
本发明中,所述催化氢化的条件没有特别的限定,可以为本领域的常规选择,例如,所述催化氢化反应的条件包括:温度15-30℃,时间为6-12h;为了提高催化氢化反应的效率,优选地,所述催化氢化反应的条件包括:温度20-30℃,时间8-10h。
[0186]
通过上述方法,可以制备得到本发明第一方面所述的多羟基吡咯烷类化合物。
[0187]
本发明第三方面提供一种糖苷酶抑制剂,所述糖苷酶抑制剂含有本发明第一方面所述的多羟基吡咯烷类化合物或本发明第二方面所述方法制备得到的多羟基吡咯烷类化合物。
[0188]
根据本发明第三方面,所述糖苷酶抑制剂用于抑制α-葡萄糖苷酶、β-葡萄糖苷酶、α-半乳糖苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-甘露糖苷酶、α-l-岩藻糖苷酶、α,α-海藻糖酶、α-l-鼠李糖苷酶、淀粉葡糖苷酶和β-葡糖苷酸化酶中的一种或多种。
[0189]
本发明中,所述糖苷酶抑制剂特别是对于α-葡萄糖苷酶、β-葡萄糖苷酶和β-半乳糖苷酶具有十分良好的抑制活性。
[0190]
另外,本发明式(1-1)至式(1-20)所示化合物对上述糖苷酶表现出良好的抑制效果。
[0191]
因此,在本发明一个特别优选的实施方式中,本发明还提供上述式(1-1)至式(1-20)所示化合物中的一种或多种作为糖苷酶抑制剂的应用。
[0192]
特别优选地,本发明提供上述式(1-1)至式(1-20)所示化合物中的一种或多种作为糖苷酶抑制剂在抑制α-葡萄糖苷酶、β-葡萄糖苷酶和β-半乳糖苷酶中的应用。
[0193]
本发明第四方面提供本发明第一方面所述的多羟基吡咯烷类化合物或本发明第二方面所述方法制备得到的多羟基吡咯烷类化合物在制备药物中的应用,所述药物选自下述药物中的至少一种:1)预防和/或治疗糖尿病的药物;2)预防和/或治疗高雪氏病的药物;3)预防和/或治疗溶酶体贮积症的药物;4)预防和/或治疗肿瘤的药物;5)抗病毒药物。
[0194]
根据本发明,在应用本发明所述的多羟基吡咯烷类化合物制备所述药物时,所述药物中还可以加入一种或多种药学上可接受的载体。例如,所述载体可以包括药学领域通常使用的各种稀释剂、赋形剂、填充剂、粘合剂、湿润剂、崩解剂、吸收促进剂、表面活性剂、吸附载体和润滑剂中的一种或多种,没有特别的限定。
[0195]
根据本发明,所述药物可以制备成注射液、片剂、粉剂、颗粒剂、胶囊、口服液、膏剂和霜剂等多种本领域常规的形式,没有特别的限定。
[0196]
另外,根据本发明,应用本发明所述的多羟基吡咯烷类化合物制备得到的药物可以利用各种给药途径给药,包括但不限于口服、吸入、直肠、透皮、经黏膜内给药,以及经皮下、肌肉或静脉注射给药等多种形式。
[0197]
以下将通过实施例对本发明进行详细描述。
[0198]
以下制备例和测试例中,如无特殊说明,所述方法均为本领域常规方法,所用试剂均为商业途径获得的试剂。
[0199]
以下制备例中采用的硝酮(2-1)和(2-2)均购自百灵威科技有限公司。
[0200]
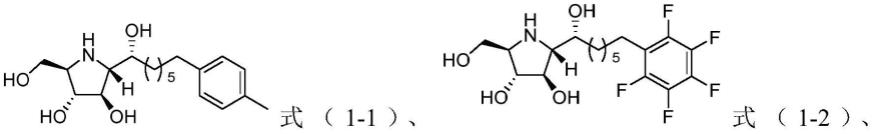
制备例1:式(1-1)所示化合物的制备
[0201][0202]
1)将式(2-1)所示硝酮(6.280g,15mmol)溶于50ml干燥的thf(四氢呋喃)中,在氩气保护下,在0℃下将乙烯基溴化镁溶液(30mmol)缓慢注入其中,待滴加完毕后,在室温(25℃,下同)下反应30min,之后加入饱和氯化铵溶液淬灭反应,用乙酸乙酯萃取水相3次,合并有机相,用饱和氯化钠溶液洗涤后,加入无水mgso4干燥,过滤后减压蒸馏除去溶剂,得到6.312g的粗产物,即式(3-1)所示羟胺;
[0203]
2)在20℃下,将上述粗产物(15mmol)溶于50ml的醋酸中,之后加入cu(oac)2(0.264g,1.5mmol)和活化锌粉(14.625g,0.225mol,经1mol/l的盐酸活化),室温下反应4h,反应完毕后,锌粉过滤后用乙酸乙酯反复冲洗锌粉,将滤液旋干,之后用乙酸乙酯溶解,再加入饱和碳酸氢钠水溶液进行中和后,用乙酸乙酯萃取水相3次,接着合并有机相,加入无水mgso4干燥,过滤后除去溶剂,得到6.301g羟胺还原的粗产物;
[0204]
将上述羟胺还原的粗产物(15mmol)溶于50ml的thf中,加10ml的水和碳酸氢钠(2.52g,30mmol),接着缓慢滴加cbzcl(3.839g,22.5mmol),在室温下搅拌1h后去除溶剂,加入乙酸乙酯与水溶解所得产物后,用乙酸乙酯洗涤水相3次,之后合并有机相,用无水mgso4干燥后旋干溶剂,经过柱层析分离得到式(4-1)所示产物6.521g,从式(2-1)所示硝酮出发至此步骤的总产率为77%;
[0205]
3)将上述产物,即式(4-1)所示化合物(2.748g,4.878mmol)溶于50ml二氯甲烷中,加入间氯过氧苯甲酸(1.684g,9.757mmol)和碳酸氢钠(0.820g,9.757mmol),在室温下反应3天,反应后加入过量的na2s2o3溶液淬灭反应,接着用二氯甲烷萃取水相3次,合并有机相后旋干,经过柱层析分离得式(5-1)所示化合物(1.191g,2.054mmol,产率42%)和式(6-1)所示化合物(0.502g,0.8660mmol,产率17%);
[0206][0207]
4)将镁屑(0.234g,9.73mmol)加入两口瓶中,在氩气保护下加入20ml干燥的thf,之后缓慢注入5-溴-1-戊烯(1.208g,8.11mmol),用一粒碘(约50mg)引发反应,加热到45℃反应30min,得到5-溴-1-戊烯的格氏试剂(mgbr-ch
2-(ch2)
2-ch=ch2)备用;
[0208]
将步骤3)得到的式(5-1)所示化合物(2.352g,4.054mmol)溶于50ml干燥的thf中,加入cui(77mg,0.405mmol)后,在室温下缓慢注入上述制备得到的5-溴-1-戊烯的格氏试剂(6.2mmol),反应40min后,缓慢滴加饱和nh4cl水溶液淬灭反应。接着加入乙酸乙酯萃取水相,合并有机相并旋干后,经过柱层析分离得到式(7-1)所示化合物2.163g,此步骤的产率为82%;
[0209]
5)将步骤4)所得产物,即式(7-1)所示化合物(128mg,0.197mmol)溶于干燥的二氯甲烷(5ml)中,加入4-甲基苯乙烯(362mg,0.985mmol)和催化剂hoveyda-grubbs ii(20mg,0.002mmol),在氩气保护下反应过夜(大约8h),反应完毕后,旋干溶剂,经过柱层析分离即可得到式(9-1)所示化合物110mg,此步骤的产率为75%;
[0210]
6)将步骤5)所得产物,即式(9-1)所示化合物(110mg,0.148mmol)溶于10ml甲醇中,加入hcl溶液(1ml,1mol/l)和pd/c(10mg)后,在氢气气氛下反应过夜(大约8h),之后滤除pd/c,将溶剂旋干即可得到50mg式(1-1)所示化合物,产率100%。
[0211]1h nmr(400m,cd3od)δ7.09
–
7.00(m,4h),4.23(t,j=6.9hz,1h),3.97(dd,j1=6.9hz,j2=8.3hz,1h),3.90
–
3.76(m,3h),3.43
–
3.37(m,2h),2.55(t,j=7.52hz,2h),2.27(s,3h),1.66
–
1.57(m,4h),1.54
–
1.46(m,1h).1.43
–
1.31(m,5h)
13
c nmr(100m,cd3od)δ139.4,134.6,128.5,127.9,75.2,73.7,68.6,64.6,63.4,57.3,35.1,33.2,31.3,29.1,28.8,25.4,19.7;ir(neat,kbr,cm-1
)3386,2926,2854,1729,1652,1647,1069.
[0212]
制备例2:式(1-3)所示化合物的制备
[0213][0214]
按照制备例1所述方法进行,不同的是,
[0215]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的2,3,4,5,6-五氟苯乙烯,最终得到41mg式(1-2)所示化合物,产率为62%。
[0216]1h nmr(500m,cd3od)δ4.24(t,j=7hz,1h,),3.98(t,j=8.1hz,1h),3.93
–
3.78(m,3h),3.41(t,j=4.2hz,2h),2.75
–
2.62(m,2h),1.66
–
1.56(m,4h);1.56
–
1.48(m,1h),1.48
–
1.34(m,5h)
13
c nmr(125m,cd3od)δ146.2
–
134.2(m),113.9(t,j=19.2hz),75.2,73.7,68.54,64.5,63.4,57.3,33.1,29.3,29.0,28.9,28.8,28.7,25.32,25.3,21.7,21.4.
[0217]
制备例3:式(1-3)所示化合物的制备
[0218][0219]
按照制备例1所述方法进行,不同的是,
[0220]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氯苯乙烯,最终得到42mg式(1-3)所示化合物。
[0221]1h nmr(400m,cd3od)δ7.26(t,j=7.4hz,2h),7.19
–
7.13(m,2h),4.26(t,j=7.0hz,1h),3.99(t,j=8.2hz,1h),3.92
–
3.78(m,3h),3.45
–
3.38(m,2h),2.63(t,j=7.5hz,2h),1.71
–
1.61(m,4h),1.60
–
1.48(m,1h),1.48
–
1.35(m,5h);
13
c nmr(100m,cd3od)δ142.5,128.0,127.9,125.3,75.2,73.7,68.6,64.6,63.4,57.3,35.5,33.2,31.3,29.1,28.8,25.4.
[0222]
制备例4:式(1-4)所示化合物的制备
[0223][0224]
按照制备例1所述方法进行,不同的是,
[0225]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-叔丁基苯乙烯,最终得到52mg式(1-4)所示化合物。
[0226]1h nmr(500m,cd3od)δ7.28(d,j=8.1hz,2h),7.09(d,j=8.1hz,2h),4.25(t,j=7.2hz,1h),4.00(t,j=6.8hz,1h),3.94
–
3.79(m,3h),3.42(dd,j=2hz,j=4.4hz,2h),2.57(t,j=7.5hz,2h),1.68
–
1.59(m,4h),1.57
–
1.48(m,1h),1.45
–
1.36(m,5h),1.30(s,9h);
13
c nmr(125m,cd3od)δ148.0,139.4,127.7,124.7,75.2,73.7,68.6,64.5,63.4,57.3,35.0,33.8,33.2,31.3,30.6,29.1,28.9,25.4.
[0227]
制备例5:式(1-5)所示化合物的制备
[0228][0229]
按照制备例1所述方法进行,不同的是,
[0230]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氟苯乙烯,最终得到55mg式(1-5)所示化合物。
[0231]1h nmr(500m,cd3od)δ7.16(dd,j1=5.8hz,j2=8.4hz,2h),6.96(t,j=8.8hz,2h),4.05(t,j=6.4hz,1h),3.82(t,j=6.6hz,1h),3.73
–
3.58(m,3h),3.08(dd,j1=6.3hz,j2=10.7hz,1h),2.99(dd,j1=4.5hz,j2=6.7hz,1h),2.59(t,j=7.6hz,2h),1.64
–
1.56(m,3h),1.56
–
1.46(m,2h),1.43
–
1.32(m,5h);
13
c nmr(125m,cd3od)δ161.2(d.j=240hz,),138.4,129.6(d,j=7.7hz),114.4(d,j=21.1hz),75.2,73.6,68.6,64.5,63.4,57.2,34.6,33.2,31.3,29.0,28.7,25.3.
[0232]
制备例6:式(1-6)所示化合物的制备
[0233][0234]
按照制备例1所述方法进行,不同的是,
[0235]
步骤4)中,将式(5-1)所示化合物替换为相同摩尔量的式(6-1)所示化合物,最终得到41mg式(1-6)所示化合物。
[0236]1h nmr(500m,cd3od)δ7.09
–
6.99(m,4h),4.24(t,j=7.0hz,1h),3.98(dd,j=6.9hz,j=8.4hz,1h),3.90
–
3.75(m,3h),3.40(dt,j=5.0hz,j=9,2hz,2h),2.57(t,j=7.5hz,2h),2.29(s,3h),1.67
–
1.55(m,4h),1.55
–
1.45(m,1h),1.45
–
1.32(m,5h);
13
c nmr(125m,cd3od)δ139.4,134.6,128.5,127.9,75.2,73.6,68.6,64.5,63.4,57.2,35.0,33.2,31.3,29.1,28.8,25.4,19.7,19.65.
[0237]
制备例7:式(1-7)所示化合物的制备
[0238][0239]
按照制备例6所述方法进行,不同的是,
[0240]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的2,3,4,5,6-五氟苯乙烯,最终得到39mg式(1-7)所示化合物。
[0241]1h nmr(500m,cd3od)δ4.24(t,j=7hz,1h,),3.98(t,j=8.1hz,1h),3.92
–
3.87(m,1h),3.86(dd,j=3.6hz,j=12.2hz,1h),3.84(dd,j=5.2hz,j=11.9hz,1h)3.43
–
3.37(m,2h),2.75(t,j=7.4hz,2h),1.66
–
1.59(m,4h);1.55
–
1.48(m,1h),1.44
–
1.36(m,5h)
13
c nmr(125m,cd3od)δ145.6
–
115.5(m),75.2,73.7,68.5,64.5,63.4,57.3,33.1,28.9,28.8,28.7,25.3,21.7.
[0242]
制备例8:式(1-8)所示化合物的制备
[0243][0244]
按照制备例6所述方法进行,不同的是,
[0245]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氯苯乙烯,最终得到55mg式(1-8)所示化合物。
[0246]1h nmr(500m,cd3od)δ7.28(d,j=4.4hz,2h),7.19
–
7.13(m,2h),4.23(t,j=6.4hz,1h),3.97(t,j=6.8hz,1h),3.90
–
3.75(m,3h),3.43
–
3.35(m,2h),2.61(t,j=5.5hz,2h),1.66
–
1.57(m,4h),1.54
–
1.46(m,1h),1.43
–
1.33(m,5h);
13
c nmr(125m,cd3od)δ142.5,128.0,127.9,125.3,75.2,73.7,68.6,64.6,63.4,57.3,35.5,33.2,31.3,29.1,28.8,25.4.
[0247]
制备例9:式(1-9)所示化合物的制备
[0248][0249]
按照制备例6所述方法进行,不同的是,
[0250]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-叔丁基苯乙烯,最终得到53mg式(1-9)所示化合物。
[0251]1h nmr(500m,cd3od)δ7.27(d,j=8.2hz,2h),7.07(d,j=8.2hz,2h),4.23(t,j=7.0hz,1h),3.96(dd,j=6.8hz,j=8.4hz,1h),3.90
–
3.85(m,1h),3.81(dq,j=5.3hz,j=12hz,2h),3.41
–
3.36(m,2h),2.56(t,j=7.5hz,2h),1.65
–
1.58(m,4h),1.54
–
1.46(m,1h),
1.43
–
1.34(m,5h),1.29(s,9h);
13
c nmr(125m,cd3od)δ148.0,139.4,127.7,124.7,75.2,73.7,68.6,64.5,63.4,57.2,35.0,33.7,33.2,31.3,30.6,29.1,28.9,25.4.
[0252]
制备例10:式(1-10)所示化合物的制备
[0253][0254]
按照制备例6所述方法进行,不同的是,
[0255]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氟苯乙烯,最终得到55mg式(1-10)所示化合物。
[0256]1h nmr(500m,cd3od)δ7.17(dd,j1=5.7hz,j2=8.4hz,2h),6.97(t,j=8.8hz,2h),4.24(t,j=7.1hz,1h),3.98(dd,j=6.9hz,j=8.4hz,1h),3.90
–
3.86(m,1h),3.82(dq,j=3.5hz,j=12.2hz,2h)3.42
–
3.37(m,2h),2.60(t,j=7.6hz,2h),1.66
–
1.59(m,4h),1.53
–
1.46(m,1h),1.43
–
1.32(m,5h);
13
c nmr(125m,cd3od)δ161.2(d.j=240.2hz,),138.4(d,j=3.2hz),129.5(d,j=7.7hz),114.4(d,j=21.2hz),75.2,73.7,68.6,64.5,63.4,57.2,34.6,33.2,31.3,29.0,28.7,25.3.
[0257]
制备例11:式(1-11)所示化合物的制备
[0258][0259]
按照制备例1所述方法进行,不同的是,
[0260]
步骤1)中,将式(2-1)所示硝酮替换为相同摩尔量的式(2-2)所示硝酮,最终得到55mg式(1-11)所示化合物。
[0261]1h nmr(500m,cd3od)δ7.06(dd,j1=2.4hz,j2=8.5hz 4h),4.02(dt,j1=6.4hz,j2=11.6hz 2h),3.91
–
3.80(m,3h),3.40(d,j=2.9hz 1h),3.25(t,j=5.8hz 1h),2.57(t,j=7.5hz,2h),2.29(s,3h),1.66
–
1.48(m,6h),1.45
–
1.35(m,4h);
13
c nmr(125m,cd3od)δ139.4,134.6,128.5,127.9,75.9,74.7,67.5,66.3,63.6,57.5,35.0,34.0,31.3,29.0,28.8,25.1,19.7.
[0262]
制备例12:式(1-12)所示化合物的制备
[0263][0264]
按照制备例11所述方法进行,不同的是,
[0265]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的2,3,4,5,6-五氟苯乙烯,最终得到36mg式(1-12)所示化合物。
[0266]1h nmr(500m,cd3od)δ4.01
–
3.95(m,2h),3.87
–
3.75(m,4h),3.21
–
3.15(m,1h),
2.72(t,j=7.2hz,2h),1.65
–
1.52(m,5h),1.45
–
1.38(m,5h);
13
c nmr(125m,cd3od)δ76.4,75.2,68.0,66.2,63.5,58.2,34.0,29.1,28.8,28.7,25.0,21.7.
[0267]
制备例13:式(1-13)所示化合物的制备
[0268][0269]
按照制备例11所述方法进行,不同的是,
[0270]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氯苯乙烯,最终得到44mg式(1-13)所示化合物。
[0271]1h nmr(500m,cd3od)δ7.23(t,j=7.6hz,2h),7.17
–
7.11(m,2h),4.01(dt,j=6.4hz,j=9.7hz,2h),3.89
–
3.80(m,3h),3.39(t,j=3hz,1h),3.25(t,j=5.1hz,1h),2.60(t,j=7.5hz,2h),1.67
–
1.49(m,6h),1.44
–
1.35(m,4h);
13
c nmr(125m,cd3od)δ142.5,128.0,127.9,125.2,75.9,74.7,67.5,66.4,63.6,57.5,37.6,35.5,34.1,33.2,26.4,26.1,25.1.
[0272]
制备例14:式(1-14)所示化合物的制备
[0273][0274]
按照制备例11所述方法进行,不同的是,
[0275]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-叔丁基苯乙烯,最终得到57mg式(1-14)所示化合物。
[0276]1h nmr(500m,cd3od)δ7.17(d,j=8.2hz,2h),6.98(d,j=8.2hz,2h),3.90
–
3.85(m,2h),3.77
–
3.67(m,3h),3.26
–
3.22(m,1h),3.11
–
3.07(m,1h),2.47(t,j=7.6hz,2h),1.57
–
1.39(m,6h),1.38
–
1.25(m,4h),1.19(s,9h);
13
c nmr(125m,cd3od)δ148.0,139.4,127.7,124.7,76.3,75.1,67.8,66.2,63.5,58.0,34.9,33.7,31.3,30.5,29.0,28.9,25.1.
[0277]
制备例15:式(1-15)所示化合物的制备
[0278][0279]
按照制备例11所述方法进行,不同的是,
[0280]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氟苯乙烯,最终得到55g式(1-15)所示化合物。
[0281]1h nmr(500m,cd3od)δ7.17(dd,j=5.6hz,j=8.4hz,2h),6.97(t,j=8.8hz,2h),4.0
–
3.94(m,2h),3.90
–
3.74(m,3h),3.35
–
3.33(m,1h),3.21
–
3.14(m,1h),2.60(t,j=7.6hz,2h),1.69
–
1.47(m,6h),1.42
–
1.32(m,4h);
13
c nmr(125m,cd3od)δ162.2(d,j=240.3hz),138.4,129.5(d,j=7.5hz),114.4(d,j=20.9hz),76.4,75.1,67.9,66.2,63.5,58.1,34.6,34.1,31.3,29.0,28.7,25.1.
[0282]
制备例16:式(1-16)所示化合物的制备
[0283][0284]
按照制备例11所述方法进行,不同的是,
[0285]
步骤4)中,将式(5-2)所示化合物替换为相同摩尔量的式(6-2)所示化合物,最终得到46mg式(1-16)所示化合物。
[0286]1h nmr(400m,cd3od)δ7.09
–
7.02(m,4h),4.10
–
3.96(m,2h),3.95
–
3.82(m,3h),3.47
–
3.39(m,1h),3.31
–
3.25(m,1h),2.57(t,j=7.6hz,2h),2.29(s,3h),1.66
–
1.51(m,5h),1.47
–
1.34(m,5h);
13
c nmr(125m,cd3od)δ139.4,134.6,128.5,127.9,75.9,74.7,67.5,66.3,63.6,57.5,35.0,34.0,31.3,29.0,28.8,25.1,19.7.
[0287]
制备例17:式(1-17)所示化合物的制备
[0288][0289]
按照制备例16所述方法进行,不同的是,
[0290]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的2,3,4,5,6-五氟苯乙烯,最终得到31mg式(1-17)所示化合物。
[0291]1h nmr(500m,cd3od)δ4.04
–
3.97(m,2h),3.88
–
3.81(m,3h),3.42
–
3.35(m,1h),3.26
–
3.19(m,1h),2.74(t,j=7.4hz,2h),1.65
–
1.52(m,5h),1.45
–
1.38(m,5h);
13
c nmr(125m,cd3od)δ76.0,74.8,67.7,66.3,63.5,57.5,35.5,33.9,28.9,28.7,25.0,21.7.
[0292]
制备例18:式(1-18)所示化合物的制备
[0293][0294]
按照制备例16所述方法进行,不同的是,
[0295]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氯苯乙烯,最终得到48mg式(1-18)所示化合物。
[0296]1h nmr(500m,cd3od)δ7.25(t,j=7.6hz,2h),7.17
–
7.11(m,2h),4.03(dt,j=6.4hz,j=9.7hz,2h),3.93
–
3.80(m,3h),3.44
–
3.40(m,1h),3.27(t,j=5.1hz,1h),2.62(t,j=7.5hz,2h),1.70
–
1.51(m,6h),1.40
–
1.39(m,4h);
13
c nmr(125m,cd3od)δ142.5,128.0,127.9,125.2,75.9,74.7,67.5,66.4,63.6,57.5,37.6,35.5,34.1,33.2,26.4,26.1,25.1.
[0297]
制备例19:式(1-19)所示化合物的制备
[0298][0299]
按照制备例16所述方法进行,不同的是,
[0300]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-叔丁基苯乙烯,最终得到51mg式(1-19)所示化合物。
[0301]1h nmr(500m,cd3od)δ7.28(d,j=8.2hz,2h),7.09(d,j=8.2hz,2h),4.06
–
3.99(m,2h),3.91
–
3.81(m,3h),3.43
–
3.38(m,1h),3.26(t,j=5.2hz,1h),2.58(t,j=7.6hz,2h),1.68
–
1.50(m,5h),1.38
–
1.36(m,5h),1.31(s,9h);
13
c nmr(125m,cd3od)δ148.0,139.4,127.7,124.7,75.9,74.7,67.6,66.3,63.6,57.6,35.0,33.8,31.3,30.6,29.0,28.9,25.1.
[0302]
制备例20:式(1-20)所示化合物的制备
[0303][0304]
按照制备例16所述方法进行,不同的是,
[0305]
步骤5)中,将4-甲基苯乙烯替换为相同摩尔量的4-氟苯乙烯,最终得到47mg式(1-20)所示化合物。
[0306]1h nmr(500m,cd3od)δ7.16(t,j=7.1hz,2h),6.96(t,j=8.4hz,2h),4.50(q,j=5.9hz,2h),4.10(t,j=5.6hz,1h),3.79(t,j=6.8,1h),3.75(dd,j=3.8hz,j=11.7hz,1h),3.67(dd,j=4.1hz,j=11.6hz,1h),3.56(q,j=4.1hz,1h),3.47(t,j=6.0hz,1h),2.59(t,j=7.4hz,2h),1.85
–
1.75(m,1h),1.75
–
1.67(m,1h),1.66
–
1.57(m,2h)1.53
–
1.27(m,6h);
13
c nmr(125m,cd3od)δ161.2(d,j=240.1hz),138.4(d,j=3.3hz),129.5(d,j=7.5hz),114.4(d,j=21.3hz),80.8,80.1,78.5,67.7,65.9,60.9,34.9,34.6,31.3,27.7,27.6,24.1.
[0307]
测试例
[0308]
(1)试验材料及来源
[0309]
供试化合物:本发明制备例提供的式(1-1)至式(1-20)所示的多羟基吡咯烷类化合物。
[0310]
试验材料:4-硝基酚吡喃糖苷基质、二糖和各种糖苷酶(包括α-葡萄糖苷酶、β-葡萄糖苷酶、α-半乳糖苷酶、β-半乳糖苷酶、α-甘露糖苷酶、β-甘露糖苷酶、α-l-岩藻糖苷酶、α,α-海藻糖酶、α-l-鼠李糖苷酶、淀粉葡糖苷酶以及β-葡糖苷酸化酶)均购自sigma-aldrich。
[0311]
(2)试验方法
[0312]
动力学研究在37℃的柠檬酸钠/磷酸缓冲液(50mmol/l)中进行。根据基质的不同,配制的酶浓度为0.1-0.5mg/ml。活性测试以4-硝基酚吡喃糖苷为基质,在每种酶的最佳活性ph下进行测试。将基质、酶溶液和抑制剂(本发明的多羟基吡咯烷类化合物)在37℃下培养30分钟,然后在紫外可见分光光度计中启动反应,测定其对400nm波长光的吸收。最后使用grafit程序进行数据分析[参见leatherbarrow,r.j.grafit4.0;erithacussoftware:staines,uk,1998.]。
[0313]
(3)评价结果
[0314]
本发明提供的多羟基吡咯烷类化合物对糖苷酶的抑制活性结果见表1-4。
[0315]
下表中,ic
50
表示对酶的抑制率为50%时的抑制剂浓度;括号内的百分数表示抑制剂浓度为1000μmol/l时对酶的抑制率。
[0316]
表1
[0317][0318]
表2
[0319][0320]
表3
[0321][0322]
表4
[0323][0324]
通过上表中的数据可以看出,本发明的多羟基吡咯烷类化合物对于糖苷酶具有良好的抑制活性。尤其是对β-葡萄糖苷酶具有优异的抑制活性,同时还体现出良好的β-葡萄糖苷酶选择性。
[0325]
此外,在多种来源的β-葡萄糖苷酶中,本发明提供的多羟基吡咯烷类化合物尤其是对于牛肝来源的β-葡萄糖苷酶表现出非常优异的抑制活性。
[0326]
通过本发明提供的测试例可以看出,本发明制备例制备得到的式(1-1)所示化合物和式(1-3)所示化合物显示出了非常显著的对于牛肝来源的β-葡萄糖苷酶抑制活性,与其它化合物相比抑制活性提高了十倍以上,甚至可以高达近千倍。与此同时,式(1-1)所示化合物和式(1-3)所示化合物还表现出明显更低的对大鼠小肠麦芽糖酶来源的α-葡萄糖苷酶的抑制活性,由此表明式(1-1)所示化合物和式(1-3)所示化合物作为药物组分摄入后对肠道的影响显著降低,从而会大大降低产生腹泻等副作用的几率和程度。十分适合作为药
物组分应用。
[0327]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1