一种ω-转氨酶突变体及其在制备西塞卡纳药物中间体的应用
一种
ω-转氨酶突变体及其在制备西塞卡纳药物中间体的应用
技术领域
1.本发明涉及分子生物学技术领域,具体涉及一种ω-转氨酶突变体及其在制备西塞卡纳药物中间体的应用。
背景技术:
2.手性胺由于其高密度的氢键的形成和精细的生物活性,逐渐成为许多医药领域的关键组成部分药物。手性胺经常用于合成手性药物,如阿尔茨海默病药物卡巴拉汀、肾上腺素能拮抗剂地来洛尔、抗逆转录病毒药物洛匹那韦、糖尿病药物西他列汀等。这些手性化合物的应用药物极大地促进了高效合成药物的探索手性胺。
3.(r)-1-(1-萘基)乙胺(r-nea)是一种重要的手性芳香胺,是重要的医药中间体和中间体手性拆分剂。例如,r-nea和3-(3-三氟甲基苯基)-丙酸可以进行酰胺偶联用于生产拟钙剂盐酸西那卡塞用于治疗与甲状旁腺癌相关的继发性甲状旁腺功能亢进和高钙血症。此外,(r)-和(s)-1-(1-萘基)乙胺可吸附在聚结晶铂表面,制备高活性手性催化剂。与传统的化学合成方法相比,生物催化是光活性胺合成的绿色替代方法。生物催化剂通常在环境温度和中性ph下运行在水介质中,催化手性中间体的生物合成高效可行的方式。在过去几十年里,各种已经探索了用于手性胺合成的酶,如胺脱氢酶、单胺氧化酶、还原性氨基酶、亚胺还原酶、氨解酶和转氨酶。
4.转氨酶是催化某基团从供体化合物转移到受体化合物上的一类酶,可以通过简单的催化剂里,可逆地将来源于氨基酸供体的氨基转移到氨基酸受体上。目前,在不对称合成手性胺类化合物及胺类化合物外消旋体拆分中,转氨酶都是关键的生物技术酶。由于其不对称催化合成手性胺的特点,转氨酶已成为工业上用于生产氨基酸、手性胺、氨基醇和氨基糖等重要农药或医药中间体的常用酶之一。来自于土曲霉属(aspergillus terreus)的ω-转氨酶以酮类化合物为原料,通过立体选择性地转氨基作用,可以高效生产手性胺,催化氨基供体上的氨基转移到前手性的受体酮,得到手性胺和副产物酮,反应过程需要磷酸吡哆醛(pyridoxal phosphate,plp)的参与,实验表明,ω-转氨酶野生型在40℃下的半衰期仅为6.9min,不利于应用到工业生产中,其热稳定性有待进一步提高。如cn105441404a、cn105950581a公开了利用定点突变技术对ω-转氨酶野生型进行改造,获得热稳定性进一步提高的ω-转氨酶突变体,使其更适合工业应用。
5.转氨酶在合成手性胺方面具有较好的应用前景,但由于野生型酶在底物特异性、稳定性、催化效率等方面存在诸多不足,目前满足工业应用需求的转氨酶仍较为有限。基于非理性、理性及半理性设计策略的蛋白质工程技术能有效改善转氨酶的应用性能,为手性胺的高效制备提供了可能。随着转氨酶蛋白结构和催化机制相关研究的深入,利用理性或半理性设计策略对转氨酶进行分子改造的研究备受关注。
6.目前尚无利用转氨酶催化口袋疏水氨基酸位阻效应结合对硝基苯乙胺的显色反应,进而利用定点突变技术进行改造方法来提高土曲霉属(aspergillus terreus)ω-转氨
酶热稳定性的相关研究报道。
技术实现要素:
7.本发明提供了一种ω-转氨酶突变体及其在制备西塞卡纳药物中间体的应用,旨在筛选出热稳定性和酶活极大提高的土曲霉(aspergillus terreus)ω-转氨酶突变体。
8.本发明基于一种高通量显色反应定量检测的方法,通过该方法通过颜色差异,紫外吸收强度以及高效液相色谱检测并用,能够快速准确筛选高产(r)-1-(1-萘基)乙胺的土曲霉(aspergillus terreus)ω-转氨酶突变体。本发明利用转氨酶催化口袋疏水氨基酸位阻效应,筛选与野生酶催化口袋附近的疏水氨基酸残基,作为优选突变氨基酸,进而利用定点突变技术进行改造。同时,结合对硝基苯乙胺的显色反应,对转氨酶催化生产西塞卡纳药物中间体(r)-1-(1-萘基)乙胺的进行定量分析。
9.本发明提供了一种ω-转氨酶突变体,由来自土曲霉(aspergillus terreus)的ω-转氨酶突变所得,野生型ω-转氨酶的氨基酸序列如seq id no.2所示,所述ω-转氨酶突变体的突变位点为:v149a/r128l/l182v/d224k、v149a/r128l/l182f/d224k或v149a/l182f/l187f/d224k中的一种。该ω-转氨酶以1-乙酰基萘作为底物、所述ω-转氨酶突变体为催化剂,存在氨基供体1-(r)-苯乙胺的条件下,合成(r)-1-(1-萘基)乙胺。
10.本发明还提供了编码上述ω-转氨酶突变体的基因。优选的,突变位点为v149a/r128l/l182v/d224k、v149a/r128l/l182f/d224k或v149a/l182f/l187f/d224k的ω-转氨酶突变体的基因序列分别如seq id no.3-5所示。
11.本发明还提供了包含上述基因的重组表达质粒。本发明还提供了包含上述重组表达质粒的基因工程菌。本发明还提供了上述的ω-转氨酶突变体、基因或基因工程菌在催化1-乙酰基萘生成西塞卡纳药物中间体(r)-1-(1-萘基)乙胺中的应用。相较于野生型酶,突变体酶在较高温条件下具有较好的热力学稳定性,更适合工业应用。
12.本发明还提供了一种催化1-乙酰基萘生成西塞卡纳药物中间体(r)-1-(1-萘基)乙胺的方法,1-乙酰基萘作为氨基受体,1-(r)-苯乙胺作为氨基供体,使用上述ω-转氨酶突变体或基因工程菌进行催化反应,将来自氨基供体的氨基转至氨基受体,反应得到西塞卡纳药物中间体(r)-1-(1-萘基)乙胺。催化反应时,所述1-乙酰基萘的摩尔浓度为10-50mm,所述1-(r)-苯乙胺的摩尔浓度为10-50mm;催化反应的温度为30℃;催化反应的时间为0.5-24h。
13.可选的,所述at-ata突变体由重组质粒pet-28a+-at-ata表达。该重组质粒的原始载体为pet-28a+。
14.本发明基于一种催化口袋疏水氨基酸的位阻效应,确定需要突变的氨基酸残基位点,结合高通量显色的定量筛选方法,通过定点突变技术进行实验验证。生物催化生成(r)-1-(1-萘基)乙胺(西塞卡纳药物中间体),与其他化合物反应生可成化合物西塞卡纳。相对于化学合成法,利用ω-转氨酶生物催化合成上述手性胺化合物,合成条件温和,操作简单,易分离提取。该方法可有效地提高突变氨基酸位点的筛选概率,提升高通量筛选的效率,增加了高通量筛选的精度,提高实验效率及可行性,并筛选得到热力学稳定性、酶活性明显优于野生酶的突变酶。
附图说明
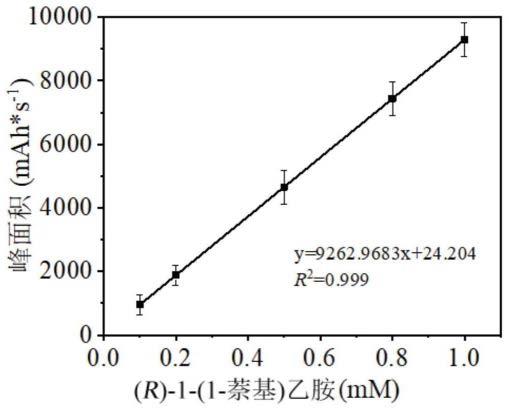
15.图1为(r)-1-(1-萘基)乙胺的定量显色筛选标准曲线图。
16.图2为实施例1中不同蛋白浓度下,(r)-1-(1-萘基)乙胺的生成量与δod
500
的线形图和96孔板显色图。
17.图3为实施例1中不同底物浓度下的96孔板显色图。
18.图4为实施例1中不同菌体浓度下,(r)-1-(1-萘基)乙胺的生成量与δod
500
的线形图。
19.图5为实施例1中制备得到的转氨酶野生型及其突变体m1(v149a/r128l/l182v/d224k)、m2(v149a/r128l/l182t/d224k)和m3(v149a/l182f/l187f/d224k)的sds-page分析结果图。
20.图6为转氨酶野生型和突变体m3的热力学性质图;其中,a:野生型与突变体m3的最适温度曲线图;b:野生型与突变体m3的半衰温度图;c:野生型与突变体m3在40℃下的半衰时间图。
21.图7为转氨酶野生型和突变型在不同反应条件下(实施例2~实施例6)催化底物1-乙酰基萘生成(r)-1-(1-萘基)乙胺的催化效率分析图;其中,a:ω-转氨酶及突变体对1-乙酰基萘的比活力对比图;b:在不同浓度的1-(r)-苯乙胺下,ω-转氨酶及突变体m3的催化效率图;c:在不同浓度的1-乙酰基萘下ω-转氨酶及突变体m3的催化效率图。
22.图8为转氨酶野生型和突变型在不同反应条件下(实施例2~实施例6)催化底物1-乙酰基萘生成(r)-1-(1-萘基)乙胺的催化效率分析图;其中,a:ω-转氨酶及突变体全细胞催化10mm底物浓度的转化率图;b:ω-转氨酶及突变体全细胞催化20mm底物浓度的转化率图;c:ω-转氨酶及突变体全细胞催化30mm底物浓度的转化率图。
23.图9为(r)-1-(1-萘基)乙胺标准品的高效液相色谱图。
24.图10为ω-转氨酶比活力测定生成(r)-1-(1-萘基)乙胺的高效液相色谱图。
25.图11为ω-转氨酶突变体酶比活力测定生成(r)-1-(1-萘基)乙胺的高效液相色谱图。
26.图12为野生转氨酶全细胞催化24h所得反应液高效液相色谱图。
27.图13为突变体m1全细胞催化24h所得反应液高效液相色谱图。
28.图14为突变体m2全细胞催化24h所得反应液高效液相色谱图。
29.图15为突变体m3全细胞催化24h所得反应液高效液相色谱图。
30.图16为fdaa标准品高效液相色谱图
31.图17为fdaa-1-(r)-苯乙胺标准品高效液相色谱图。
32.图18为fdaa-1-(s)-苯乙胺标准品高效液相色谱图。
33.图19为fdaa-(r)-1-(1-萘基)乙胺标准品高效液相色谱图。
34.图20为fdaa-(s)-1-(1-萘基)乙胺标准品高效液相色谱图。
35.图21为野生转氨酶全细胞催化24h所得反应液的衍生物高效液相色谱图。
36.图22为突变体m3全细胞催化24h所得反应液的衍生物高效液相色谱图。
具体实施方式
37.实施例1
38.1、实验材料
39.(1)lb培养基:10g/l胰蛋白胨(购于oxoid),5g/l酵母粉(购于oxoid),10g/l氯化钠(购于生工生物工程有限公司(上海)),ph 7.0。lb固体培养基:lb液体培养基加入2%(质量比)琼脂粉。
40.(2)二甲基亚砜(dmso)和na-(2,4-二硝基-5-氟苯基)-l-丙氨酰胺购于阿拉丁试剂有限公司。甘氨酸、tris、十二烷基硫酸钠、通用型蛋白染色液、氯化钠、甘油、氯化钙、咪唑、冰醋酸、磷酸氢二钠、磷酸二氢钠、5-磷酸吡哆醛、考马斯亮蓝蛋白质浓度测定试剂盒、ni-nta层析介质、异丙基-β-d-硫代半乳糖苷(iptg)、溶菌酶、硫酸卡那霉素和磷酸吡哆醛(pyridoxal-5-phosphate,plp)购于上海生工生物工程有限公司。对硝基苯乙胺、1-乙酰基萘、1-(r)-苯乙胺、(r)-1-(1-萘基)乙胺和(s)-1-(1-萘基)乙胺购于上海毕得医药科技股份有限公司。dna marker、蛋白质marker、蛋白上样缓冲液和核酸染料购于北京全式金生物技术有限公司。琼脂粉和无水乙醇购于国药集团化学试剂有限公司。乙腈购于上海沃凯化学试剂有限公司。
41.(3)菌株:本技术中经密码子优化的ω-转氨酶基因(at-ata基因)(如seq id no.1所示)委托通用生物系统(安徽)有限公司进行全基因合成,基因合成服务中使用pet-28a+质粒作为克隆载体,酶切位点分别为ndei和hindiii。构建的重组质粒pet-28a+-at-ata(转氨酶突变体v149a/r128l/l182v/d224k、v149a/r128l/l182f/d224k和v149a/l182f/l187f/d224k质粒,编码基因序列如seq id no.3-5所示,转入e.coli bl21(de3),获得重组菌。
42.2、(r)-1-(1-萘基)乙胺标准曲线的建立
43.配置0.1-1mm 1-乙酰基萘稀释液,用磷酸盐缓冲液(pbs,50mm,ph 8.0)进行稀释,以pbs作为对照组,用50%乙腈:水溶液(v/v)对稀释液稀释后进行液相分析。标准曲线图如图1所示。
44.3、显色反应体系的构建及优化
45.(1)纯酶催化体系的显色反应
46.分别配置不同纯酶浓度的200μl反应液,其中包括终浓度分别为0.005-0.1mg/ml的纯酶,12.5mm 4-硝基苯乙胺,5mm 1-乙酰基萘,体积比为20%的dmso以及0.1mm plp,用pbs进行稀释。以磷酸盐缓冲液作为对照,观察1小时内反应溶液的颜色变化,并取100μl最终反应液,用50%乙腈:水溶液(v/v)稀释后进行液相分析。不同蛋白浓度下,(r)-1-(1-萘基)乙胺的生成量与δod500的线形图和96孔板显色图如图2所示。
47.分别配置不同底物浓度的200μl反应液,其中包括终浓度为0.1mg/ml的纯酶,12.5-100mm 4-硝基苯乙胺,5-40mm 1-乙酰基萘,氨基供体:氨基受体投料比为4,体积比为20%的dmso以及0.1mm plp,用磷酸盐缓冲液(pbs,50mmol/l,ph8.0)进行稀释。以磷酸盐缓冲液作为对照,观察1小时内反应溶液的颜色变化,并取100μl最终反应液,用50%乙腈:水溶液(v/v)稀释后进行液相分析。不同底物浓度下的96孔板显色图如图3所示。
48.(2)全细胞催化体系的显色反应
49.分别取5-25mg的沉菌溶液,用pbs稀释至50μl,加入150μl的底物溶液,其中包括25mm 4-硝基苯乙胺,10mm 1-乙酰基萘,体积比为20%的dmso以及0.1mm plp,用pbs进行稀释。以pbs作为对照,混匀后测定0-30min内反应溶液od
500
。取100μl反应30min之后的反应液,用50%乙腈:水溶液(v/v)稀释后进行液相分析,并计算δod
500
,建立其与峰面积的线性
关系。不同菌体浓度下,(r)-1-(1-萘基)乙胺的生成量与δod
500
的线形图如图4所示。
50.4、高效液相hplc检测方法的建立
51.高效液相色谱条件(用于产物检测):色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.03%乙醇胺),b相(乙腈)。流动相采用等度洗脱程序:0-15min,a相的比例为60%。
52.产物衍生条件:参照文献(r.bhushan and h.bruckner.marfey’s reagent for chiral amino acid analysis:a review.aminoacids,2004,27(3):231-247.)的方法进行衍生反应。将50μl反应液与100μl1%marley丙酮稀释液(m/v)混合,加入20μlnahco3溶液(ph 9.8),置于40℃、400rpm反应2h,用20μl hcl溶液(2m)淬灭反应。用3倍体积的二氯甲烷进行萃取,常温挥发之后,溶解于50%乙腈(v/v):水溶液并稀释,进行液相分析。
53.高效液相色谱条件(用于手性检测):色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.1%甲酸),b相(乙腈)。流动相采用等度洗脱程序:0-8min,a相的比例为30%。
54.5、固相筛选方法的构建
55.(1)重组质粒的转化
56.取出-80℃保藏的e.coli bl21(de3)感受态细胞,冰上解冻后加入2μl pet-28a+-at-ata稀释液(20ng/μl),混合后置于冰上30min。于42℃水浴中热激90s后,迅速置于冰上充分冷却5min。向管中加入650μl lb液体培养基,37℃、180rpm培养1.5h,使大肠杆菌复性,并表达重组质粒编码的卡那霉素抗性基因。取100μl菌液稀释均匀涂布于含有50μg/ml的卡那霉素的lb平板上,正面向上放置半小时,待菌液完全被培养基吸收后倒置平板,37℃培养24h后放置于4℃冰箱保存。
57.(2)固相显色反应的构建
58.从上述平板上挑取单菌落接种至5ml含50μg/ml卡那霉素的lb培养基中,37℃、200rpm条件下培养6h。取100μl菌液稀释适当倍数后均匀涂布于含有50μg/ml的卡那霉素的lb平板上的n66尼龙微孔滤膜上,37℃静置培养12h。用镊子将长有单菌落的微孔滤膜转移至含有0.5mm iptg和50μg/ml的卡那霉素的lb平板上,25℃静置培养8h诱导目的蛋白at-ata的产生。将长有诱导后单菌落的微孔滤膜置于含有显色反应液(25mm 4-硝基苯乙胺,10mm 1-乙酰基萘,体积比为20%的dmso以及0.1mm plp,用pbs进行稀释)的滤纸上,30℃反应30min,观察菌落的颜色变化。以对照组a和对照组b进行对照实验,其中对照组a为没有进行诱导的单菌落进行固相显色反应,对照组b为长有经过诱导的单菌落的微孔滤膜置于不含1-乙酰基萘的显色反应液的滤纸上进行固相显色反应。
59.6、突变位点的选择
60.在不考虑表面氨基酸与1-乙酰基萘存在相互作用的前提下,将催化口袋附近的疏水氨基酸和相互作用氨基酸进行筛选,得到三个备选突变为位点(v149、l182和l187)进行饱和突变。使用表1中的引物、以含有ω-转氨酶的编码基因(ω-转氨酶的氨基酸序列如seq id no.2所示,基因序列如seq id no.1所示,长度为978bp,从土曲霉aspergillus terreus中克隆所得,genebank number:mz855772)的质粒为模板进行pcr扩增。在37℃条件下,pcr产物经dpn i酶进行消化后,进行pcr产物纯化试剂盒进行纯化,之后采用化学转化法转入e.coli dh 5α感受态细胞中,1h后将复苏液涂布于含有终浓度为50μg/μl卡那霉素的lb固
体平板得到定点突变文库。重组质粒送至安徽通用生物系统有限公司进行核苷酸序列测定,将测序正确的重组质粒转化入e.coli bl21(de3)感受态细胞,以获取目标重组菌株。
61.at-ata突变体v149a/r128l/l182v/d224k(命名为突变体m1)、v149a/r128l/l182f/d224k(命名为突变体m2)、v149a/l182f/l187f/d224k(命名为突变体m3)对应的各位点饱和突变和定点突变引物如表1所示。
62.表1
63.突变位点序列r128x-fttaaaggggtnnkaggaactcgtccggaagatr128x-ratatcttccggacgagttcctmnnacccctttv147xv149x-ftagtgaacaacctgtacatgtttgtgcagccgtacnnktggnnkatggagccggatatv147xv149x-rtccggctccatmnnccamnngtacggctgcacaaacatgtacaggttgttcactatatl182x-ftattgatccgaccgtcaagaatnnkcagtggggtgatcttgttl182x-raacaagatcaccccactmnngattcttgacggtcggatcaatal187x-fgaatcttcagtggggtgatnnkgttcgtggaatgtttgaagl187x-rgcttcaaacattccacgaacmnnatcaccccactgaagattcd224k-fttagtcaaaaaaggcgtcctgtatacgccagatd224k-rtggcgtatacaggacgcctttttgactaatac
64.7、突变文库的建立与质粒的提取
65.取出-80℃保存的e.coli dh5α感受态细胞并将其放冰上解冻。5μl的消化产物加入到50μl e.coli dh5α感受态细胞中,用枪轻轻混匀,冰上静置30min。42℃水浴锅内热激90s,迅速将管放冰上冷却3~5min。每管中加入600μl预冷的lb培养基,37℃,180rpm条件下复苏培养1h,使细菌恢复到正常生长状态。将菌液6000rpm离心2min,除去500μl上清液,取余下菌液混匀后,均匀地涂布于含有终浓度为50μg/ml kan的lb固体培养基平板上。37℃培养箱中培养平板(正面向上)20-30min后,倒置平板,过夜培养。
66.随机挑取平板上的单菌落接种于5ml含50μg/ml kan的lb培养基中,37℃,230rpm条件下培养至od
600
值为0.8时,取1ml菌液送至杭州擎科梓熙生物技术有限公司进行核苷酸序列的测定,1ml菌液用于保存菌种,3ml菌液用于提取质粒,具体步骤参照质粒小量提取试剂盒说明书。测序正确的菌种质粒用1%dna琼脂糖凝胶电泳验证目的条带的大小和纯度后,5μl质粒被转入e.coli bl21(de3)感受态细胞中,以获取目标重组菌株。
67.8、酶的表达与纯化
68.挑取野生型及突变体单菌落接种于5ml含有终浓度为50μg/ml kan的lb液体培养基中,37℃、230rpm条件下摇床培养12h。菌液以2%的接种量(v/v)转接至200ml含有终浓度为50μg/ml kan的lb液体培养基中,37℃、230rpm条件下继续培养2~3h。当od
600
达到0.8时,加入终浓度为0.5mm的iptg,并在25℃、150rpm条件下诱导蛋白表达。诱导20h后,在6000rpm,4℃条件下收集菌体。
69.菌体细胞用20mm的pbs缓冲液(ph 8.0)洗涤2次去除残留培养基后悬浮于50ml破胞缓冲液(50mm磷酸二氢钠,300mm氯化钠,20mm咪唑,ph8.0)中。冰浴条件下对菌体细胞进行均质机破碎细胞。细胞破碎液在8000rpm,4℃条件下离心1h,收集得到的上清液即为含有ω-转氨酶的粗酶液。随后,采用ni-nta亲和层析法对粗酶液进行分离纯化,粗酶经上样、清
洗、洗脱后得到纯酶液,操作步骤参照说明书进行。
70.9、蛋白含量的测定
71.采用改良型bradford蛋白浓度测定试剂盒建立蛋白含量标准曲线,测定纯酶的浓度,蛋白标准曲线的制备步骤参照说明书进行。采用sds-page方法(12%分离胶和5%浓缩胶)鉴定纯化后蛋白的分子量和纯度。
72.野生型和突变体的sds-page电泳图谱如图5所示。野生型和突变体的电泳条带位于同一水平线,分子量约为36kda,与理论分子量(36.1kda)相一致,为后续实验的进行奠定了基础。
73.10、最适温度的测定方法
74.酶的最适温度的测定方法如下:在500μl的底物溶液中,包含10mm 1-(r)-苯乙胺,10mm 1-乙酰基萘,体积百分比为20%的dmso,0.1mm plp以及0.02mg/ml at-ata野生型和突变体m3纯酶液,用ph8.0的pbs缓冲液进行稀释,并迅速置于不同温度(25-45℃)的恒温混匀仪中,400rpm条件下反应30min。将反应液用50%乙腈:水溶液(v/v)稀释后进行液相分析。野生型与突变体m3的最适温度曲线图如图6a所示。
75.11、半衰期
76.半衰期(t
1/2
)是指在40℃时,at-ata及其突变体的剩余活力下降至50%所需的时间。酶的最适温度的测定方法如下:将稀释为0.02mg/mlat-ata纯酶液置于40℃孵育0-30min,随后在冰上放置5min,用于配置反应体系。在500μl的底物溶液中,包含10mm 1-(r)-苯乙胺,10mm 1-乙酰基萘,体积百分比为20%的dmso,0.1mm plp以及0.02mg/ml经过预处理的at-ata纯酶液,用ph8.0的pbs缓冲液进行稀释,并迅速置于30℃的恒温混匀仪中,400rpm条件下反应30min。将反应液用50%乙腈:水溶液(v/v)稀释后进行液相分析。以温度为横坐标,以热处理后与处理前酶活力的比值为纵坐标作图,采用origin8.0的非线性回归的指数函数模型:exp2pmod1(y=exp(-kd·
t))对数据进行拟合,求出一级速率常数(kd),并计算酶的活力降低至50%时所对应的半衰期(t
1/2
)。野生型与突变体m3在40℃下的半衰时间图如图6c所示。突变体m3的半衰温度为46.5℃,比野生型提升了8.6℃。
77.12、半失活温度
78.半失活温度是指在不同温度连续加热10min后,酶的活力降至一半的温度。将纯化后的at-ata及其突变体在4、25、35、37、40、42、45、50和55℃的温度下孵育10min,然后在冰上冷却5min,采用上述方法测定酶活力。将数据拟合到用levenberg-marquardt迭代算法改进的boltzmann sigmoid函数。野生型与突变体m3的半衰温度图如图6b所示。突变体m3在40℃下孵育的半失活时间为46.9min,较野生型提高了6.3倍。
79.采用上述ω-转氨酶突变体生物催化合成(r)-1-(1-萘基)乙胺见以下实施例2~6。
80.实施例2
81.ω-转氨酶及其突变体m3催化氨基从氨基供体转移到1-乙酰基萘生成(r)-1-(1-萘基)乙胺:底物溶液用磷酸盐缓冲液(50mm,ph 8.0)配制,5ml的反应体系中包括10-50mm 1-(r)-苯乙胺,10-50mm 1-乙酰基萘,0.1mm plp,0.01-0.1mg/ml ω-转氨酶突变体,30℃下进行催化反应1h,空白对照则用缓冲溶液代替ω-转氨酶突变体。结果如图7b-c可知,野生型和突变体m3均能够利用1-乙酰基萘进行转氨反应,生成(r)-1-(1-萘基)乙胺,且产量
随着1-乙酰基萘浓度增加而增加,随着1-(r)-苯乙胺浓度增加而降低。
82.实施例3
83.ω-转氨酶及其突变体催化氨基从氨基供体转移到1-乙酰基萘生成(r)-1-(1-萘基)乙胺:底物溶液用磷酸盐缓冲液(50mm,ph 8.0)配制,500μl的反应体系中包括10mm 1-(r)-苯乙胺,10mm 1-乙酰基萘,0.1mm plp,0.1mg/ml ω-转氨酶突变体,30℃下进行催化反应15min,空白对照则用缓冲溶液代替ω-转氨酶突变体。ω-转氨酶及其突变体酶比活力测定生成(r)-1-(1-萘基)乙胺的高效液相色谱图如图10和图11所示;结果如图7a可知,野生型和突变体m1、m2、m3均能够利用1-乙酰基萘进行转氨反应,生成(r)-1-(1-萘基)乙胺,突变体m1、m2、m3的催化效率明显高于野生型。
84.实施例4
85.ω-转氨酶及其突变体催化氨基从氨基供体转移到1-乙酰基萘生成(r)-1-(1-萘基)乙胺:底物溶液用磷酸盐缓冲液(50mm,ph 8.0)配制,10ml的反应体系中包括10mm 1-(r)-苯乙胺,10mm 1-乙酰基萘,0.1mm plp,0.1g/ml产ω-转氨酶及其突变体的目的菌株,30℃下进行催化反应0.5-24h,空白对照则用缓冲溶液代替ω-转氨酶突变体。将产物稀释适当倍数进行液相分析:色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.03%乙醇胺),b相(乙腈)。流动相采用等度洗脱程序:0-15min,a相的比例为60%。
86.将产物进行衍生反应检测其手性:将50μl反应液与100μl 1%marfey丙酮稀释液(m/v)混合,加入20μl nahco3溶液(ph 9.8),置于40℃、400rpm反应2h,用20μl hcl溶液(2m)淬灭反应。用3倍体积的二氯甲烷进行萃取,常温挥发之后,溶解于50%乙腈:水溶液并稀释,进行液相分析。高效液相色谱条件:色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.1%甲酸),b相(乙腈)。流动相采用等度洗脱程序:0-8min,a相的比例为30%。结果如图8a可知,野生型和突变体m1、m2、m3均能够利用10mm 1-乙酰基萘进行转氨反应,生成(r)-1-(1-萘基)乙胺,转化率可以达到78%(投料比1∶1)。
87.实施例5
88.ω-转氨酶及其突变体催化氨基从氨基供体转移到1-乙酰基萘生成(r)-1-(1-萘基)乙胺:底物溶液用磷酸盐缓冲液(50mm,ph 8.0)配制,10ml的反应体系中包括20mm1-(r)-苯乙胺,20mm 1-乙酰基萘,0.1mm plp,0.1g/ml(r)-表达ω-转氨酶及其突变体,30℃下进行催化反应0.5-24h,空白对照则用缓冲溶液代替ω-转氨酶突变体。将产物稀释适当倍数进行液相分析:色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.03%乙醇胺),b相(乙腈)。流动相采用等度洗脱程序:0-15min,a相的比例为60%。
89.将产物进行衍生反应检测其手性:将50μl反应液与100μl 1%marfey丙酮稀释液(m/v)混合,加入20μl nahco3溶液(ph 9.8),置于40℃、400rpm反应2h,用20μl 2m hcl溶液淬灭反应。用3倍体积的二氯甲烷进行萃取,常温挥发之后,溶解于50%乙腈:水溶液并稀释,进行液相分析。高效液相色谱条件:色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.1%甲酸),b相(乙腈)。流动相采用等度洗脱程序:0-8min,a相的比例为30%。结果如图8b可知,野生型和突变
体m1、m2、m3均能够利用20mm 1-乙酰基萘进行转氨反应,生成(r)-1-(1-萘基)乙胺,转化率可以达到60%(投料比1∶1)。
90.实施例6
91.ω-转氨酶及其突变体催化氨基从氨基供体转移到1-乙酰基萘生成(r)-1-(1-萘基)乙胺:底物溶液用磷酸盐缓冲液(50mm,ph8.0)配制,10ml的反应体系中包括30mm1-(r)-苯乙胺,30mm 1-乙酰基萘,0.1mm plp,0.1g/ml(r)-表达ω-转氨酶及其突变体,30℃下进行催化反应0.5-24h,空白对照则用缓冲溶液代替ω-转氨酶突变体。将产物稀释适当倍数进行液相分析,ω-转氨酶及其突变体全细胞催化24h所得反应液高效液相色谱图如图12-15所示。
92.将产物进行衍生反应检测其手性:将50μl反应液与100μl 1%marfey丙酮稀释液(m/v)混合,加入20μl nahco3溶液(ph 9.8),置于40℃、400rpm反应2h,用20μl hcl溶液(2m)淬灭反应。用3倍体积的二氯甲烷进行萃取,常温挥发之后,溶解于50%乙腈:水溶液并稀释,进行液相分析。野生转氨酶和突变体m3全细胞催化24h所得反应液的衍生物高效液相色谱图如图21和图22所示;结果如图8c可知,野生型和突变体m1、m2、m3均能够利用30mm 1-乙酰基萘进行转氨反应,生成(r)-1-(1-萘基)乙胺,转化率可以达到46%(投料比1∶1)。
93.实施例2-6反应产物的hplc分析方法及结果:
94.产物高效液相色谱条件:色谱柱为agilent infinitylab poroshell 120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.03%乙醇胺),b相(乙腈)。流动相采用等度洗脱程序:0-15min,a相的比例为60%。
95.手性高效液相色谱条件:色谱柱为agilent infinitylab poroshell120(2.1
×
100mm,1.8μm),柱温30℃;进样量为10μl;流动相为:a相(水+0.1%甲酸),b相(乙腈)。流动相采用等度洗脱程序:0-8min,a相的比例为30%。
96.如图9所示,产物(r)-1-(1-萘基)乙胺的液相出峰时间为5.703min,峰形单一尖锐,无明显杂峰;反应物手性衍生物液相出峰时间为4.242min(fdaa-1-(r)-苯乙胺,标准品标准品高效液相色谱图如图17所示)和4.704min(fdaa-1-(s)-苯乙胺,标准品标准品高效液相色谱图如图18所示),峰形单一尖锐,无明显杂峰;产物手性衍生物液相出峰时间为4.705min(fdaa-(r)-1-(1-萘基)乙胺,标准品标准品高效液相色谱图如图19所示)和5.409min(fdaa-(s)-1-(1-萘基)乙胺,标准品标准品高效液相色谱图如图20所示)峰形单一尖锐,无明显杂峰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1