一种拉诺康唑的制备方法与流程
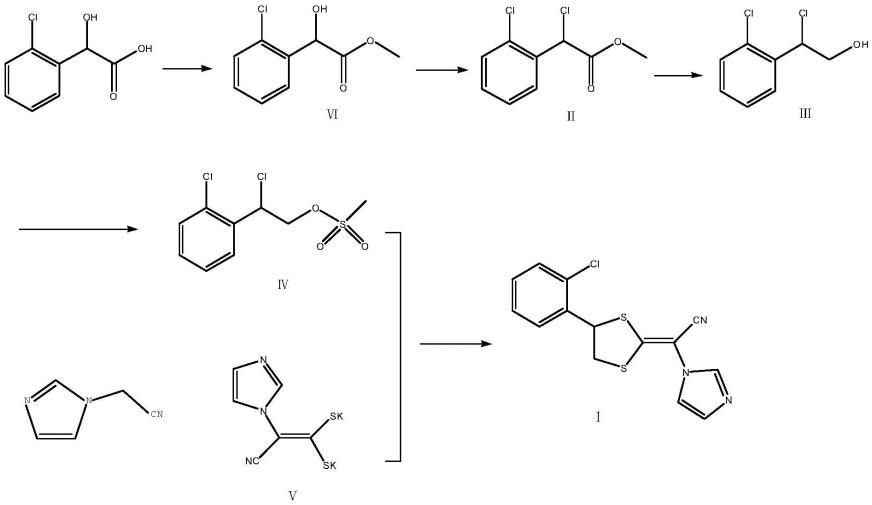
1.本发明涉及一种拉诺康唑的制备方法,属于药物合成技术领域。
背景技术:
2.拉诺康唑(lanoconazole,1)是日本tsumura株式会社开发,1994年首先在日本上市的新型、强效咪唑类抗真菌药物,其商品名为
[0003][0004]
拉诺康唑作为新一代抗真菌药物,其作用机制同其他咪唑类药物,主要通过抑制14-甲基羊毛甾醇的脱14-甲基阶段,达到抑制真菌细胞麦角固醇合成的目的,从而具有抗真菌作用。在临床上,对于广泛致病真菌:包括酵母菌、皮肤真菌、真菌、二形真菌、曲霉菌属、青霉菌属和念珠菌属均具有很强的抗菌活性,尤其是对毛癣菌属特别有效。用于真菌感染疾患包括足癣、体癣、股癣、念珠菌病、花斑癣等皮肤真菌病的治疗,效果非常明显。
[0005]
拉诺康唑是咪唑类中的新一代抗真菌药物,其临床疗效、副作用表现,均优于其他的抗真菌药物,有着明显的竞争优势,必将成为抗真菌药的畅销品种,具有较好的社会经济效益。
[0006]
现有合成拉诺康唑的文献报道的合成路线经环合反应得到含z和e异构体的混合物,都需要使用柱层析的方法,才能得到拉诺康唑(e-式构型)。如jp02275877、us4636519制得反应液浓缩物,通过柱层析,分离纯化,得到e异构体总收率20~28%,副产物z异构体的占比为20~31%。又如中国专利(公开号:cn101701017a)提供的拉诺康唑的合成路线,通过合成关键中间体1-(2-氯苯基)-1,2-乙二醇二甲磺酸酯,与1-咪唑基乙腈、二硫化碳和氢氧化钾反应得到的二硫醇盐环合反应得到z/e异构体混合物,经柱层析分离纯化,制备最终产物拉诺康唑。该反应过程中形成中间体1-(2-氯苯基)-1,2-乙二醇二甲磺酸酯的化学性质太过活泼,在反应过程中极易发生水解,使后续的环合反应不易实现,也造成产物收率低及生成的杂质多等问题,且该反应在环合反应后同样无法实现z/e构形的选择性,仍需通过柱层柱进行分离,存在难分离和不易操作等问题。
技术实现要素:
[0007]
本发明针对以上现有技术中存在的缺陷,提供一种拉诺康唑的制备方法,解决了现有反应不稳定和收率低的问题。
[0008]
本发明的目的是通过以下技术方案得以实现的,一种拉诺康唑的制备方法,以2-氯扁桃酸为原料先经过酯化反应和氯代反应合成中间体式ⅱ化合物,其特征在于,该方法还包括以下步骤:
[0009]
a、在还原剂硼氢化物的存在下,使式ⅱ化合物在水溶性有机溶剂中进行还原反
应,得到中间体式ⅲ化合物;
[0010][0011]
b、在敷酸剂存在下,在非水溶性溶剂中使式ⅲ化合物与甲磺酰氯进行反应,得到中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯;
[0012][0013]
c、在有机溶剂和水的混合液中加入1-咪唑基乙腈、二碳化碳和相转移催化剂,加入强碱性的钾盐进行反应,得到含有式
ⅴ
化合物的反应液,向反应液中加入弱碱性的缓冲体系,向所述反应液中加入中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯进行环合反应,反应结束后,浓缩除去溶剂,再加入良性溶剂进行重结晶,得到相应的产物式ⅰ化合物拉诺康唑;
[0014][0015]
本发明通过以2-氯扁桃酸为原料,先经过氯代反应将其中的羟基进行氯代后再进行酯还原和磺酰化反应形成本方法的关键中间体1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯,相当于使该中间体的中α-位的羟基以氯进行取代后,有效的避免该位羟基存在时太过活泼的问题,有效避免因存在两个羟基时发生水解的问题,且通过氯取代后形成的中间体能够使其与甲磺酰氯进行反应时能够温和进行,避免杂质过多的产生,有利于提高中间体及最终产物的收率和纯度质量;同时,在后续的环合反应过程中能够有效的形成e构形的拉诺康唑产物,具有反应稳定性好的优点;且使反应在弱碱性的缓冲体系下进行,能够得到化学热力学稳定的产物拉诺康唑(e-式),减少副产物z构型结构,反应液中的副产物z型的异构体含量在10%以下,远远低于现有常规的方法,从而实现无需采用柱层析进行分离和纯化,再用大量hbr转构型,本发明只需要通过重结晶进行产物的分离纯化即可,大大的简化了操作,且具有产物收率高和优点,也避免了大量的高沸点、强酸性废液的产生,减少对环境的污染。
[0016]
在上述拉诺康唑的制备方法中,作为优选,步骤b中所述敷酸剂选自三乙胺、二乙胺、吡啶和哌啶中的一种或几种。目的在于除去反应过程中形成的小分子酸,使有利于反应进行和中间体的转化,提高中间体的产物收率。步骤b中所述敷酸剂的用量优选为式ⅲ化合物与敷酸剂的摩尔比为1:1.0~1.3。作为更进一步的优选,步骤b中所述非水溶性溶剂选自
二氯甲烷、二氯乙烷、乙酸乙酯、醋酸异丙酯和甲苯中的一种或几种。能够更好的使反应温和进行,还有利于该步反应的后处理。进一步的讲,通过在反应结束后,还可加入水进行洗涤,搅拌静置,更有利于除去水溶性杂质,提高中间体的纯度质量,而中间体萃取到有机层中也能够有效的分离得到。上述对于溶剂的用量可根据一般的用量均可。更进一步的优选,步骤b中所述反应的温度为10℃~20℃。通过采用本发明合成的中间体式ⅲ化合物与甲磺酰氯反应,能够使反应温和进行,且使反应温度在较低的状态下进行,也能够更好的减少杂质的产生,提高中间体的质量。
[0017]
在上述拉诺康唑的制备方法中,作为优选,步骤c中所述弱碱性的缓冲体系选自磷酸氢二盐(钠或钾)-磷酸二氢盐(钠或钾)缓冲体系、碳酸盐(钠或钾)-碳酸氢盐(钠或钾)缓冲体系。磷酸二氢盐或碳酸盐的用量为式iv化合物的质量用量的13%~15%,磷酸氢二盐或碳酸氢盐的用量为式iv化合物的质量用量的11%~15%,能够使反应在相对稳定的ph值(8~9)体系下进行,提高了反应的整体稳定性,使形成的产物是化学热力学稳定的e-式构型的拉诺康唑,具有产物纯度高的优点。上述的磷酸氢二盐(钠或钾)-磷酸二氢盐(钠或钾)缓冲体系,如可以是磷酸氢二钠-磷酸氢钠缓冲体系,或磷酸氢二钾-磷酸二氢钾缓冲体系;上述的碳酸盐(钠或钾)-碳酸氢盐(钠或钾)缓冲体系,如可以是碳酸钠-碳酸氢钠缓冲体系,碳酸钾-碳酸氢钾缓冲体系等。
[0018]
在上述拉诺康唑的制备方法中,作为优选,步骤c中所述相转移催化剂选自四丁基氯化铵、四丁基溴化铵、聚乙二醇400、聚乙二醇600、18-冠-6-醚、苄基三乙基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵和十四烷基三甲基氯化铵中的一种或几种。能够更有利于反应的进行,提高产物的转化率,更进一步优选,上述相转移催化剂的用量为式iv化合物的质量用量的5%~7%。
[0019]
在上述拉诺康唑的制备方法中,作为优选,步骤c中所述良性溶剂选自选自甲醇、乙醇、异丙醇、丙酮、2-丁酮和任一它们的水溶液中的一种或几种,进一步的还可使步骤c中所述有机溶剂选自四氢呋喃、二甲基亚砜、n,n-二甲基甲酰胺、甲苯和乙酸乙酯中的一种或几种。任一它们的水溶液如乙醇水溶液、甲醇水溶液或异丙醇水溶液等。采用上述良性溶剂目的在于使有效的除去杂质和提高重结晶的质量,提高产物的收率和纯度的优点。上述对于良性溶剂的用量可根据重结晶的用量进行一般性的调整均可。
[0020]
在上述拉诺康唑的制备方法中,作为优选,步骤c中所述环合反应的温度为10℃~40℃;所述强碱性的钾盐选自碳酸钾或氢氧化钾。能够使反应温和进行,提高中间体的转化率和纯度质量。
[0021]
在上述拉诺康唑的制备方法中,作为优选,步骤a中所述硼氢化物选自硼氢化钾、硼氢化钠、硼氢化锂和硼氢化锌中的一种或几种;步骤a中所述水溶性有机溶剂选自c
1-c3的醇溶剂或c
1-c3的醇溶剂的水溶液,如甲醇、乙醇、丙醇或异丙醇,也可以是乙醇水溶液或异丙醇水溶液等。能使酯还原到醇羟基,且转化率高的优点。作为进一步的优选,最好使所述硼氢化物的摩尔用量为2-氯扁桃酸的摩尔用量的1.0~5.0,最好为2.0~3.0。
[0022]
在上述拉诺康唑的制备方法中,作为优选,步骤b中所述甲磺酰氯的摩尔用量为式ⅲ化合物的摩尔用量的1.0~1.4,最好为1.05~1.15。有利于充分利用原料,减少原料的浪费,也更有利于减少杂质的产生。
[0023]
在上述拉诺康唑的制备方法中,作为优选,步骤a中所述还原反应的温度在-5℃~
10℃,优选温度为0~5℃。更进步的优选,在还原反应结束之后,还包括向反应液中加入酸如盐酸进行淬灭,调节体系的ph值至中性,蒸馏温度控制在50℃~60℃除去部分溶剂,过滤除去体系中的固体盐,再后处理使析出需要的中间体产物,有利于提高该中间产物的纯度质量。
[0024]
在上述拉诺康唑的制备方法中,作为优选,所述酯化反应和氯代反应最好通过以下方法得到:
[0025]
在浓硫酸的作用下,将2-氯扁桃酸加入甲醇溶剂中进行酯化反应,得到中间体式ⅵ化合物;
[0026][0027]
在碱性催化剂的作用下,将中间体式ⅵ化合物与氯代试剂进行氯代反应,得到中间体式ⅱ化合物;
[0028][0029]
通过氯代反应先将α-位的羟基进行取代,使后续形成的中间体的反应活性更合适,有利于在后续的甲磺酰氯反应的稳定性,实现有效减少其它杂质产生的效果。
[0030]
上述的2-氯扁桃酸的结构式如下:
[0031][0032]
上述的酯化反应最好是在回流状态下进行,保证反应充分进行,而浓硫酸作为催化剂,采用催化量的浓硫酸起催化作用即可,最好这里的浓硫酸用量为原料2-氯扁桃酸的质量的2.0%~3.0%,促进这里的2-氯扁桃酸中的羧酸基与甲醇中的羟基进行酯化形成相应的酯基团。酯化反应结束后,可加入碱液调节体系的ph值至中性左右,再蒸馏除去溶剂,得到相应的中间体式ⅵ化合物,呈淡黄色的液体粘稠物,可直接用于下步反应;
[0033]
上述的氯代反应中氯化试剂选自草酰氯、三光气、二氯亚砜、三氯异氰脲酸、三氯化磷或五氯化磷。优选采用草酰氯或三光气。这里的碱性催化剂选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n,n-二甲基苯胺、三乙胺、咪唑和吡啶中的一种或几种,最好使所述碱性催化剂采用n,n-二甲基甲酰胺。
[0034]
本发明的拉诺康唑的制备方法的具体合成方法,可通过以下化学反应方程式进行表示:
[0035][0036]
综上所述,本发明与现有技术相比,具有以下优点:
[0037]
1.通过使先形成关键中间体关键中间体1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯,使该中间体的中α-位的羟基以氯进行取代后,能够有效的避免该位羟基存在时太过活泼的问题,有效避免发生水解的问题,且能够使其与甲磺酰氯进行反应时能够温和进行,避免杂质过多的产生,有利于提高中间体及最终产物的收率和纯度质量,具体高收率的效果,总收率达到50%~60%。
[0038]
2、最终环合反应体系下能够更有效的保证反应稳定的形成e构型的拉诺康唑,而减少副产物z构型的产生,使后处理过程中无需采用柱层析分离,只需要通过重结晶即可实现,淘汰原产能低的制备方法,实现大规模生产,具有可操作性高的优点,且废液产生少,减少对环境的污染。
具体实施方式
[0039]
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
[0040]
实施例1
[0041][0042]
在反应釜中抽入150
±
5kg无水甲醇,开启搅拌,再加入2-氯扁桃酸20kg(107.2mol)和0.5kg浓硫酸,打开热水阀门,升温至65℃进行回流反应3h,反应结束后,冷却降温至室温后,再向反应液中滴加质量百分数为10%的氢氧化钠水溶液调ph值至近中性,稳定之后,再升温至50℃~60℃进行减压蒸馏回收甲醇溶剂至干,向剩余物中补加20
±
2kg二氯甲烷,搅拌混合无均匀,得到含化合物ⅵ的二氯甲烷的淡黄色的液体粘稠物,直接进行下一步操作。
[0043]
实施例2
[0044][0045]
向反应釜抽入200
±
5kg的二氯甲烷溶剂,开启搅拌,将上述实施例1得到的化合物ⅵ溶解完全,加入0.4kg的碱性催化剂dmf,搅拌下升温至40~45℃,滴加预先加入洁净、干燥的高位槽中的草酰氯16.3kg(128.6mol)和20
±
2kg二氯甲烷的混合溶液,滴加过程控制温度在该范围,滴加结束后,继续回流反应1h,反应结束后,将反应液进行缓慢冷却降温至室温,再向反应釜中慢慢加入冷水50
±
5kg,再用质量百分数为20%的na2co3水溶液70
±
5kg,调节反应液的ph值至6~7,再搅拌30分钟中,静置分层,收集有机相,上层水相可再用50
±
5kg二氯甲烷提取一次收集有机相,将两次收集的有机相合并,将得到的有机相进行常压蒸馏回收溶剂,蒸馏至无明显溶剂蒸出时,切换至减压蒸馏至干,得中间体化合物ⅱ,呈淡黄色液体,可直接用于下步参与反应。
[0046]
实施例3
[0047][0048]
向上述实施例2得到的黄色液体化合物ⅱ中加入200
±
5kg甲醇溶剂,进行冷却降温,将反应液体系降温至0~5℃,再分批、多次加入硼氢化钾总量为14.4kg(268mol),控制在2~3h加料结束,再冰浴冷却下控制温度在0~5℃继续反应2h,反应结束后,使体系温度至室温下继续搅拌反应2h,结束后,再用质量百分数为10%的稀盐酸调体系ph至中性,再升温至50~60℃进行减压蒸除回收甲醇溶剂,蒸出约100
±
5kg,蒸馏结束后,用自来水进行冰却降至室温,使充分析晶,离心,除去白色固体盐,用20
±
2kg甲醇洗涤,滤液继续减压浓缩,蒸出约60
±
5kg甲醇,蒸馏后的残余物加水100
±
5kg,再用冰盐水进行冷却降温至0~5℃,搅拌析晶2h,离心,得类白色固体中间体化合物ⅲ,真空干燥得干品15.6kg的类白色固体中间体化合物ⅲ,收率76.1%。
[0049]
实施例4
[0050][0051]
向洁净的反应釜中加入200
±
5kg二氯甲烷、8.7kg(86.4mol)三乙胺,加入15kg(78.5mol)采用上述实施例3得到的化合物ⅲ,搅拌溶清,然后,将反应体系降温至10℃~20℃,控制温度在10℃~20℃之间滴加甲磺酰氯10.3kg(90.3mol),滴毕,再控制温度在10~20℃进行保温反应1h。保温毕,在10~20℃间滴加饮用水38
±
2kg,再升温至20℃~25℃搅拌20分钟后,静置20分钟,分层,收集有机层,向有机层中加入配制好的碳酸氢钠溶液(称取
碳酸氢钠7.6kg,加饮用水68kg
±
5kg,搅拌溶清),加毕后,搅拌20分钟,静置、分层,收集有机层,将收集的有机层采用常压蒸馏至温度50~55℃,再进行减压蒸馏至干,减压蒸馏控制温度在55℃以下,蒸馏毕,将体系降温至35℃~40℃,再抽入异丙醇37kg
±
5kg,然后,升温至回流(75℃~85℃)溶清后,再降温至0~5℃,保温搅拌0.5h,进行充分析晶,保温结束后,将料液放入离心机进行离心,滤饼用5kg
±
2kg异丙醇洗涤,出料得关键中间体化合物ⅵ的湿料,于50℃~60℃、干燥4~5h得19.3kg干品化合物ⅵ,收率91.4%。
[0052]
实施例5
[0053][0054]
向反应釜中抽入150
±
5kg四氢呋喃和70
±
5kg饮用水,再加3.8kg(35.2mol)1-咪唑基乙腈、2.8kg(36.1mol)二硫化碳和0.5kg四丁基氯化铵,搅拌10分钟后,降温至10℃~20℃,再控制温度滴加碳酸钾水溶液(称取碳酸钾9.3kg(67.5mol),加饮用水30
±
2kg,搅拌溶清),滴加结束后,搅拌下控制温度在10℃~20℃保温5小时,制得含有中间体化合物
ⅴ
反应液;
[0055]
再向含有中间体化合物
ⅴ
反应液中加入缓冲液(称取1.2kg磷酸氢二钠和1kg磷酸二氢钠,溶于20kg饮用水形成该缓冲液),搅拌10分钟后,再控制温度在15℃~25℃滴加预先配制好的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯的溶液(具体是称取9.0kg(33.4mol)中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯和30
±
2kg四氢呋喃溶液混合),滴加结束,保温反应1小时,保温完毕,控制温度在50℃以下,进行减压蒸馏回收四氢呋喃溶剂,蒸馏结束,降温至30℃以下,再抽入100
±
5kg乙酸乙酯和30
±
5kg饮用水,控制温度在15℃~25℃搅拌15~20分钟,静置10~15分钟后分层,收集有机层;水层可再加入30
±
2kg乙酸乙酯,搅拌10~15分钟,静置15分钟,分层,收集有机层。合并两次收集的有机层,再向有机层中加入配制好的质量百分数为10%的氯化钠水溶液,搅拌15分钟,静置15分钟,分层。收集的有机层控制温度在50℃以下,减压蒸馏回收溶剂至干,蒸馏结束,降温至30℃以下,抽入60
±
5kg异丙醇,升温至70~80℃回流,保温15~30分钟,结晶体系温度缓慢降至0~5℃后继续保温30分钟,保温结束后将料液放入离心机进行离心,滤饼用5
±
2kg异丙醇洗涤,出料,得最终产物拉诺康唑粗品7.9kg。
[0056]
实施例6
[0057]
在洁净的反应釜中抽入50
±
5kg异丙醇,再投入实施例5得到的拉诺康唑粗品整批7.9kg,活性炭0.5kg,升温至70~80℃回流,保温回流0.5小时,趁热压滤至结晶釜,用5
±
2kg异丙醇和10
±
2kg纯化水进行淋洗,收集的滤液进行缓慢降温至0~5℃,保温析晶0.5~1小时,进行离心分离,滤饼用5
±
2kg异丙醇淋洗,得拉诺康唑湿料,湿料在50℃下,干燥6~8小时后粉碎、过筛、包装得拉诺康唑成品6.2kg,为类白色结晶性粉末,总收率57.9%。
[0058]
实施例7
[0059]
向反应釜中抽入80
±
5kg二甲基亚砜,再依次投入3.8kg(35.2mol)1-咪唑基乙腈、
2.8kg(36.1mol)二硫化碳和0.5kg四丁基氯化铵,搅拌5~10分钟后,降温至15℃~20℃,再控制温度滴加预先配制的氢氧化钾溶液(氢氧化钾4.4kg(67.0mol),并加饮用水10
±
2kg,搅拌溶清,得到的氢氧化钾溶液),滴加结束,在15~25℃保温搅拌3小时,得到含有中间体化合物
ⅴ
反应液。
[0060]
再向含有中间体化合物
ⅴ
反应液加入缓冲液(称取1.2kg磷酸氢二钠和1kg磷酸二氢钠,溶于20kg饮用水),搅拌10分钟后。在控制温度在15℃~25℃滴加预先配制好的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯的溶液(具体是称取9.0kg(33.4mol)中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯(简称氯苯乙醇酯)和20
±
2kg二甲基亚砜溶液,滴加结束,控制温度在15~25℃下保温反应1小时;保温完毕,抽入100
±
5kg乙酸乙酯和120
±
5kg饮用水,在15~25℃搅拌15~20分钟,静置10~15分钟后分层,收集有机层;水层加入30
±
2kg乙酸乙酯,搅拌10~15分钟,静置10~15分钟,分层,收集有机层。合并两次收集的有机层,再向有机层中加入配制好的质量百分数为10%的氯化钠水溶液,搅拌15分钟,静置15分钟,分层。收集的有机层控制温度在50℃以下,进行减压蒸馏回收溶剂至干,蒸馏结束,降温至30℃以下,抽入60
±
5kg异丙醇,升温至70~80℃回流,保温15~30分钟。结晶体系温度降至0~5℃后继续保温30分钟。保温结束后将料液放入离心机进行离心,滤饼用5
±
2kg异丙醇洗涤,出料,得拉诺康唑粗品7.4kg。
[0061]
实施例8
[0062]
反应釜抽入40
±
5kg乙醇,投入实施例7得到的拉诺康唑粗品整批7.4kg,投入药用炭0.6kg,升温至70~80℃回流,保温回流0.5小时。趁热压滤至结晶釜,再用5
±
2kg乙醇和10
±
2kg纯化水混合溶液进行淋洗,收集的滤液进行缓慢降温至0~5℃,保温析晶1小时,保温结束后,进行离心分离,滤饼用5
±
2kg乙醇淋洗,得拉诺康唑湿料。湿料在45~50℃,干燥6~8小时后粉碎、过筛、包装得拉诺康唑成品5.6kg,白色结晶,总收率52.3%。
[0063]
实施例9
[0064][0065]
向反应釜抽入200
±
5kg的二氯甲烷溶剂,开启搅拌,将上述实施例1得到的化合物ⅵ溶解完全,加入0.5kg的碱性催化剂n,n-二甲基乙酰胺或三乙胺或咪唑,搅拌下升温至40~45℃,滴加预先加入洁净、干燥的高位槽中的草酰氯16.3kg(128.6mol)和20
±
2kg二氯甲烷的混合溶液,滴加过程控制温度在该范围,滴加结束后,继续回流反应1.5h,反应结束后,将反应液进行缓慢冷却降温至室温,再向反应釜中慢慢加入冷水50
±
5kg,再用质量百分数为20%的na2co3水溶液70
±
5kg,调节反应液的ph值至6~7,再搅拌30分钟中,静置分层,收集有机相,上层水相可再用50
±
5kg二氯甲烷提取一次收集有机相,将两次收集的有机相合并,将得到的有机相进行常压蒸馏回收溶剂,蒸馏至无明显溶剂蒸出时,切换至减压蒸馏至干,得中间体化合物ⅱ,呈淡黄色液体,可直接用于下步参与反应。
[0066]
本实施例中的上述草酰氯可采用三光气、三氯化磷或氯化亚砜代替,均能达到相当的水平。
[0067]
实施例10
[0068][0069]
向上述实施例2得到的黄色液体化合物ⅱ中加入200
±
5kg乙醇溶剂,进行冷却降温,将反应液体系降温至0~5℃,再分批、多次加入硼氢化钠总量为268mol,控制在2.5h加料结束,再冰浴冷却下控制温度在0~5℃继续反应2.5h,反应结束后,使体系温度至室温下继续搅拌反应2h,结束后,再用质量百分数为10%的稀盐酸调体系ph至中性,再升温至50~60℃进行减压蒸除回收乙醇溶剂,蒸出约110
±
5kg,蒸馏结束后,用自来水进行冰却降至室温,使充分析晶,离心,除去白色固体盐,用20
±
2kg乙醇洗涤,滤液继续减压浓缩蒸出约乙醇,蒸馏后的残余物加水100
±
5kg,再用冰盐水进行冷却降温至0~5℃,搅拌析晶2h,离心,得类白色固体中间体化合物ⅲ,真空干燥得干品16.2kg的类白色固体中间体化合物ⅲ,收率79.0%。
[0070]
本实施例中的还原剂硼氢化钠也可采用硼氧化锂或硼氢化锌代替,对于中间体化合物收率也能达到相当的水平,本步中中间体化合物ⅲ收率在78%以上。
[0071]
实施例11
[0072]
向洁净的反应釜中加入200
±
5kg乙酸乙酯、7.35kg(86.4mol)哌啶,加入15kg(78.5mol)采用上述实施例3得到的化合物ⅲ,
[0073]
搅拌溶清,然后,将反应体系降温至15℃,控制温度在10℃~15℃之间滴加甲磺酰氯12.4kg(94.2mol),滴毕,再控制温度在10~15℃进行保温反应2h。保温毕,控制温度在10~20℃间滴加饮用水40kg,再升温至25℃搅拌30分钟后,静置30分钟,分层,收集有机层,向有机层中加入配制好的碳酸氢钠溶液(称取碳酸氢钠8kg,加饮用水70kg,搅拌溶清),加毕后,搅拌30分钟,静置、分层,收集有机层,将收集的有机层采用常压蒸馏至温度50~55℃,再进行减压蒸馏至干,减压蒸馏控制温度在60℃以下,蒸馏毕,将体系降温至35℃~40℃,再抽入异丙醇40kg,然后,升温至回流(75℃~85℃)溶清后,再缓慢降温至0~5℃,保温搅拌1h,进行充分析晶,保温结束后,将料液放入离心机进行离心,滤饼用5kg
±
2kg异丙醇洗涤,出料得关键中间体化合物ⅵ的湿料,于50℃~60℃、干燥5h得19.5kg干品化合物ⅵ,收率92.3%。
[0074]
实施例12
[0075][0076]
向反应釜中抽入150
±
5kg四氢呋喃和70
±
5kg饮用水,再加3.8kg(35.2mol)1-咪唑基乙腈、2.8kg(36.1mol)二硫化碳和0.6kg苄基三乙基氯化铵,搅拌10分钟后,降温至10℃~20℃,再控制温度滴加碳酸钾水溶液(称取碳酸钾9.3kg(67.5mol),加饮用水30
±
2kg,搅拌溶清),滴加结束后,搅拌下控制温度在15℃保温5小时,制得含有中间体化合物
ⅴ
反应
液;
[0077]
再向含有中间体化合物
ⅴ
反应液中加入碳酸钠-碳酸氢钠缓冲体系(称取1.2kg碳酸钠和1.2kg碳酸氢钠,溶于20kg饮用水),搅拌10分钟后,再控制温度在20℃~25℃滴加预先配制好的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯的溶液(具体是称取9.0kg(33.4mol)实施例4得到的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯和30
±
2kg四氢呋喃溶液混合),滴加结束,保温反应1.5小时,保温完毕,控制温度在50℃以下,进行减压蒸馏回收四氢呋喃溶剂,蒸馏结束,降温至30℃以下,再抽入100
±
5kg乙酸乙酯和30
±
5kg饮用水,控制温度在20℃~25℃搅拌30分钟,静置30分钟后分层,收集有机层;水层可再加入30kg乙酸乙酯,搅拌15分钟,静置15分钟,分层,收集有机层。合并两次收集的有机层,再向有机层中加入配制好的质量百分数为10%的氯化钠水溶液,搅拌15分钟,静置15分钟,分层。收集的有机层控制温度在50℃以下,减压蒸馏回收溶剂至干,蒸馏结束,降温至30℃以下,抽入70kg无水乙醇,升温至70~80℃回流,保温30分钟,结晶体系温度缓慢降至0~5℃后继续保温60分钟,保温结束后将料液放入离心机进行离心,滤饼用5kg乙醇洗涤,出料,得最终产物拉诺康唑粗品8.2kg。
[0078]
向另一洁净的反应釜抽入45kg异丙醇,投入上述得到的拉诺康唑粗品整批8.2kg,投入药用活性炭0.6kg,升温至70~80℃回流,保温回流0.5小时,结束后趁热压滤至结晶釜,再用6kg异丙醇和10kg纯化水混合溶液进行淋洗,收集的滤液进行缓慢降温至0~5℃,保温析晶1.5小时,保温结束后,进行离心分离,滤饼用5kg异丙醇淋洗,得拉诺康唑湿料。湿料在45~50℃,干燥7小时后,干品进行粉碎、过筛、包装得拉诺康唑成品6.5kg,白色结晶,总收率60.7%。
[0079]
实施例13
[0080]
本实施例的拉诺康唑的制备方法同实施例5一致,区别在于将其中的相转移催化剂四丁基氯化铵分别采用四丁基溴化铵、聚乙二醇400、聚乙二醇600、18-冠-6-醚、苄基三乙基氯化铵、四丁基硫酸氢铵、十二烷基三甲基氯化铵或十四烷基三甲基氯化铵代替后一一进行实施,其它基本同实施例5一致,得到相应的最终产物拉诺康唑粗品,对应采用每个相转移催化剂时,相应产物的量均能达到7.5kg以上。表明选用的相转移催化剂具有相当的水平。
[0081]
实施例14
[0082]
本实施例的拉诺康唑的具体制备方法基本同实施例12一致,区别在于以下过程中碳酸钠-碳酸氢钠缓冲体系的加入量不同,具体如下:
[0083]
再向含有中间体化合物
ⅴ
反应液中加入碳酸钠-碳酸氢钠缓冲体系(称取1.35kg碳酸钠和1.35kg碳酸氢钠,溶于20kg饮用水),搅拌10分钟后,再控制温度在20℃~25℃滴加预先配制好的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯的溶液(具体是称取9.0kg(33.4mol)实施例4得到的中间体式ⅳ化合物1-氯-1-(2-氯苯基)-2-乙二醇甲磺酸酯和32kg四氢呋喃溶液混合),滴加结束,保温反应2.0小时,保温完毕,控制温度在50℃以下,进行减压蒸馏回收四氢呋喃溶剂,蒸馏结束,降温至30℃以下,再抽入100
±
5kg乙酸乙酯和30
±
5kg饮用水,控制温度在20℃~25℃搅拌30分钟,静置30分钟后分层,收集有机层;水层可再加入30kg乙酸乙酯,搅拌15分钟,静置15分钟,分层,收集有机层。合并两次收集的有机层,再向合并的有机层中加入配制好的质量百分数为10%的氯化钠水溶液,搅拌
15分钟,静置15分钟,分层。收集的有机层控制温度在50℃以下,减压蒸馏回收溶剂至干,蒸馏结束,降温至30℃以下,抽入70kg无水乙醇,升温至70~80℃回流,保温30分钟,结晶体系温度缓慢降至0~5℃后继续保温60分钟,保温结束后将料液放入离心机进行离心,滤饼用5kg乙醇洗涤,出料,得最终产物拉诺康唑粗品8.4kg。
[0084]
本发明中所描述的具体实施例仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
[0085]
尽管对本发明已作出了详细的说明并引证了一些具体实施例,但是对本领域熟练技术人员来说,只要不离开本发明的精神和范围可作各种变化或修正是显然的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1