吡咯酰胺化合物的制备方法与流程
1.本发明涉及药物化学领域,具体涉及吡咯酰胺化合物的制备方法,还涉及其中重要的中间体化合物及其制备方法。
背景技术:
2.wo 2006012642 a2公开了用于调节一种或多种类固醇核受体的活性的吡咯衍生物——埃沙西林酮(esaxerenone,cs-3150)的消旋化合物及其制备方法,所述化合物的结构式如下所示:
[0003][0004]
wo 2008126831 a1公开了埃沙西林酮,其可以用于治疗高血压等疾病;同时公开了其制备方法,具体过程如下所示;所述方法使用手性柱拆分而得到具有单一立体构型的目标化合物:
[0005][0006]
wo 2015030010 a1公开了用于制备埃沙西林酮化合物的中间体及其制备方法,其中所述方法通过酶拆分或光活性胺拆分以制备s构型的中间体化合物,然后与4-(甲磺酰基)苯胺在乙基溴化镁的作用下反应得到目标化合物;所述方法如方法一至方法三所示。该专利申请的方法使用价格较高的格氏试剂,所述格氏试剂用量大,且在较高温度下滴加,存在安全隐患,不利于工业化生产。此外,方法二还需使用硼氢化钠和三氟化硼-醚络合物还原羧基,该反应条件危险,也不易于放大生产。
[0007]
方法一:
[0008][0009]
方法二:
[0010][0011]
方法三:
[0012][0013]
wo 2014168103 a1公开了如下所示的另一种制备埃沙西林酮的方法,该方法通过依次制备得到s构型的羧酸和酰氯中间体化合物,然后与4-(甲磺酰基)苯胺反应以获得目标产物,但是所述酰氯中间体化合物不稳定、易变质,不利于后续反应和产品质量的控制,也不利于工业化生产。
[0014]
技术实现要素:
[0015]
为了解决上述问题,本发明提供了吡咯酰胺化合物的制备方法,其中,所述吡咯酰胺化合物包括埃沙西林酮及其类似物。同时,本发明还涉及所述方法中的重要中间体及其制备方法。本发明的制备方法条件温和,操作简单,安全可控,且产率高,产品纯度高,适合工业化生产。
[0016]
一方面,本发明提供了一种制备式(iia)化合物的方法,
[0017][0018]
其中,n为0、1、2、3或4,r和pg1具有本发明所述的含义。具体地,其制备方法如本发明所描述的方法。
[0019]
在一些实施方案中,本发明提供了一种式(ii)化合物的制备方法:
[0020][0021]
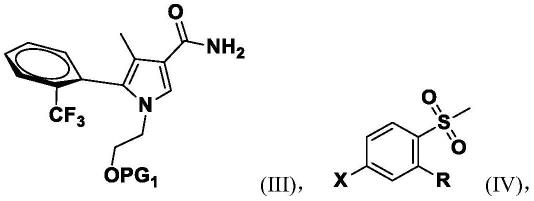
其中,所述方法包括:式(iii)化合物与式(iv)化合物反应得到式(ii)化合物;
[0022][0023]
其中,x为br或i;
[0024]
r为h、d、f、cl、br、c
1-4
烷基、c
1-4
烷氧基、c
1-4
卤代烷基或c
1-4
卤代烷氧基;
[0025]
pg1为合适的羟基保护基,包括但不限于烷基、烯基、烷氧基烷基、烷硫基烷基、硅基烷基、苄基或取代的苄基等。优选地,pg1为苄基或取代的苄基。更优选地,pg1为苄基、对甲基苄基或对甲氧基苄基。
[0026]
在另一些实施方案中,所述反应是在碱1的存在下进行的。优选地,所述碱1为碳酸钾、碳酸铯、碳酸钠、叔丁醇钾或磷酸钾。更优选地,所述碱1为碳酸钾、碳酸铯、叔丁醇钾或磷酸钾。
[0027]
在又一些实施方案中,本发明所述碱1的用量为式(iii)化合物的摩尔量的多倍,包括但不限于1.2倍至6.0倍。在又一些实施方案中,本发明所述碱1的用量为式(iii)化合物的摩尔量的1.5倍至3.0倍,优选1.5倍至2.0倍。
[0028]
在另一些实施方案中,所述反应是在催化剂的存在下进行的,所述催化剂为金属催化剂。
[0029]
在又一些实施方案中,本发明所述催化剂为钯催化剂。优选地,所述钯催化剂为pd2(dba)3、pd(dppf)cl
2.
ch2cl2或pd(oac)2。
[0030]
在又一些实施方案中,本发明所述催化剂的用量为式(iii)化合物的摩尔量的0.5%~5%,优选1%~3%,更优选1.5%~3%。
[0031]
在另一些实施方案中,本发明所述反应是在配体存在的条件下进行的。在又一些实施方案中,所述配体为dmeda、xantphos、x-phos或s-phos。在又一些实施方案中,所述配体为xantphos、x-phos或s-phos。
[0032]
在又一些实施方案中,所述配体的用量为式(iii)化合物的摩尔量的0.5%~5%,优选1%~3%,更优选1.5%~3%。
[0033]
在另一些实施方案中,本发明所述反应是在催化剂和配体的存在下进行的。
[0034]
在一些实施方案中,所述催化剂和配体的摩尔比为1:1。
[0035]
在又一些实施方案中,所述催化剂为pd(oac)2,所述配体为xantphos、s-phos或x-phos;或者,所述催化剂为pd2(dba)3,所述配体为xantphos、s-phos或x-phos;或者,所述催化剂为pd(dppf)cl
2.
ch2cl2,所述配体为xantphos、s-phos或x-phos;或者,所述催化剂为pd(dppf)cl
2.
ch2cl2,所述配体为xantphos。
[0036]
在又一些实施方案中,所述催化剂为cui,所述配体为dmeda,所述碱1为碳酸钾。
[0037]
在另一些实施方案中,所述反应是在溶剂1中进行的,所述溶剂1为甲苯、二氧六环(dioxane)、叔丁醇(t-buoh)、叔戊醇(t-amoh)、二甲醚(dme)、环戊基甲醚(cpme)、n,n-二甲基乙酰胺(dmac)、n,n-二甲基甲酰胺(dmf)、水或它们的任意组合。
[0038]
在另一些实施方案中,所述反应的反应温度为45℃-110℃,优选45℃-100℃或55℃-110℃,更优选55℃-100℃,进一步优选55℃-83℃。
[0039]
在另一些实施方案中,按照本发明所述方法制备得到的式(ii)化合物的粗产物可以通过纯化方法进行纯化。所述纯化方法包括但不限于重结晶、使用除钯硅胶和/或活性炭去除杂质等;必要时,可以进行多次重结晶。所述重结晶可以是先将粗产物溶于良性溶剂中,搅拌溶解后,加入不良溶剂或者将粗产物的溶液加入到不良溶剂中,搅拌析出固体。所述溶解过程可以是在常温条件下进行的,也可以是在加热升温的条件下进行的。优选地,在使用良性溶剂溶解粗产物后可以先使用除钯硅胶和/或活性炭等手段去除杂质,然后再将粗产物的溶液加入到不良溶剂中析出固体。在一些实施方案中,良性溶剂是乙醇,不良溶剂是水;优选地,所述乙醇和水的总的体积比可以为约1:1.5至约1:15,更优选约1:1.8至约1:15。
[0040]
在另一些实施方案中,本发明所述式(ii)化合物的制备方法还包括式(iii)化合物的制备方法,其包含如下步骤:
[0041]
步骤c:式(vi)化合物与二氯亚砜、草酰氯或n,n'-羰基二咪唑(cdi)反应得到式(v)化合物,
[0042][0043]
步骤d:式(v)化合物经氨解反应得到式(iii)化合物;
[0044]
其中,r1为cl或pg1具有本发明所述的含义。
[0045]
在又一些实施方案中,所述步骤c的反应是在0℃至室温条件下进行的。
[0046]
在又一些实施方案中,所述式(v)化合物为式(va)化合物或(vb)化合物,
[0047][0048]
在又一些实施方案中,所述步骤c中,式(vi)化合物与二氯亚砜或草酰氯反应得到式(va)化合物,
[0049][0050]
其中,所述反应是在合适的溶剂中进行的。
[0051]
在又一些实施方案中,所述合适的溶剂包括但不限于二氯甲烷(dcm)或四氢呋喃(thf)等。
[0052]
在又一些实施方案中,所述步骤c中,式(vi)化合物与n,n'-羰基二咪唑(cdi)反应得到式(vb)化合物,
[0053][0054]
其中,所述反应是在室温下于n,n-二甲基甲酰胺(dmf)或n,n-二甲基乙酰胺(dmac)等溶剂中进行的。
[0055]
在又一些实施方案中,所述步骤d的氨解反应是在氨基化试剂的存在下进行的。优选地,所述氨基化试剂为氨水或铵盐试剂;优选地,所述氨基化试剂为氨水、溴化铵或nh4scn。
[0056]
在又一些实施方案中,所述步骤d的氨解反应进一步在碱2的存在下进行,所述碱2为碳酸钾、碳酸钠或碳酸铯。
[0057]
在又一些实施方案中,所述步骤d的氨基化试剂为溴化铵或nh4scn;且所述步骤d的氨解反应在碱2的存在下进行,所述碱2为碳酸钾、碳酸钠或碳酸铯。
[0058]
在又一些实施方案中,所述步骤d的氨解反应温度为0℃至室温(如10℃至40℃)。
任选地,所述步骤d的氨解反应温度为0℃。任选地,所述步骤d的氨解反应温度为室温。
[0059]
在又一些实施方案中,所述步骤d的氨解反应得到的式(iii)化合物的粗品可经打浆或重结晶进一步纯化。
[0060]
在又一些实施方案中,所述重结晶包括:将式(iii)化合物的粗品加入溶剂a中,加热搅拌至体系溶清,加入溶剂b,冷却,搅拌析出固体。
[0061]
在又一些实施方案中,所述重结晶中,将式(iii)化合物的粗品加入溶剂a后,加热至80℃左右,搅拌至体系溶清。
[0062]
在又一些实施方案中,所述溶剂a为乙酸异丙酯或乙酸叔丁酯,溶剂b为正己烷、正庚烷或环己烷。
[0063]
在又一些实施方案中,所述溶剂a和溶剂b的体积比为1:2至4:3。
[0064]
在又一些实施方案中,所述溶剂a为乙酸异丙酯,所述溶剂b为环己烷,其中所述环己烷和乙酸异丙酯的体积比为1:1至2:1。
[0065]
在又一些实施方案中,所述溶剂a为乙酸叔丁酯,所述溶剂b为环己烷,其中所述环己烷和乙酸叔丁酯的体积比为1:1至3:4,优选3:4。
[0066]
在又一些实施方案中,以式(iii)化合物的粗品的质量计,所述溶剂a和溶剂b的总体积是6ml/g至7ml/g。
[0067]
在又一些实施方案中,本发明所述的式(iii)化合物的制备方法还包括式(vi)化合物的制备方法,其包含如下步骤:
[0068]
步骤a:式(viii)化合物与苄基溴或取代的苄基溴反应得到式(vii)化合物;
[0069][0070]
步骤b:式(vii)化合物水解得到式(vi)化合物;
[0071]
其中,r2为c
1-4
烷基、苄基或取代的苄基;
[0072]
pg1为苄基或取代的苄基。
[0073]
在一些实施方案中,所述r2为甲基、乙基、正丙基、异丙基、苄基或取代的苄基。
[0074]
在一些实施方案中,所述步骤a中的苄基溴或取代的苄基溴的摩尔量是式(viii)化合物的摩尔量的1.0倍或1.0倍以上;优选地,苄基溴或取代的苄基溴的摩尔量是式(viii)化合物的摩尔量的1.0倍至4.0倍;更优选地,苄基溴或取代的苄基溴的摩尔量是式(viii)化合物的摩尔量的1.0倍至2.5倍;进一步优选地,苄基溴或取代的苄基溴的摩尔量是式(viii)化合物的摩尔量的1.1倍至1.7倍;最优选地,苄基溴或取代的苄基溴的摩尔量是式(viii)化合物的摩尔量的1.5倍至1.7倍。
[0075]
在一些实施方案中,所述步骤a的反应是碱a的存在下进行的,所述碱a为叔戊醇钠、叔丁醇钠、乙醇钠、甲醇钠、氢氧化钠、四丁基氢氧化胺、叔戊醇钾、叔丁醇钾或氢氧化钾;优选地,所述步骤a的反应是在叔戊醇钠、乙醇钠、甲醇钠、氢氧化钠、四丁基氢氧化铵或氢氧化钾存在下进行的;更优选地,所述步骤a的反应是在叔戊醇钠、乙醇钠或氢氧化钾存
在下进行;进一步优选地,所述步骤a的反应是在叔戊醇钠存在下进行的。
[0076]
在另一些实施方案中,所述碱a的摩尔量是式(viii)化合物的摩尔量的1.0倍或以上,优选1.0倍至4.0倍,更优选1.1倍至2.5倍。
[0077]
在另一些实施方案中,所述步骤a的反应是叔戊醇钠存在下进行的,所述叔戊醇钠的摩尔量是式(viii)化合物的摩尔量的1.0倍或1.0倍以上;优选地,所述叔戊醇钠的摩尔量是式(viii)化合物的摩尔量的1.0倍至4.0倍;更优选地,所述叔戊醇钠的摩尔量是式(viii)化合物的摩尔量的1.0倍至2.5倍;进一步优选地,所述叔戊醇钠的摩尔量是式(viii)化合物的摩尔量的1.1倍至1.7倍;更优选地,所述叔戊醇钠的摩尔量是式(viii)化合物的摩尔量的1.5倍至1.7倍。
[0078]
在一些实施方案中,所述步骤a的反应可进一步在碘化钠的存在下进行。优选地,所述步骤a的反应是在催化量的碘化钠的存在下进行的;所述催化量包括但不限于0.1、0.15或0.2当量。
[0079]
在另一些实施方案中,所述碱a可以是在常温(即室温)或低温或低温至室温条件下加入的。
[0080]
在又一些实施方案中,所述步骤a的反应是叔戊醇钠存在下进行的,所述叔戊醇钠是在常温或低温或低温至室温条件下加入的;优选地,所述叔戊醇钠是在10℃至30℃条件下加入的;更优选地,所述叔戊醇钠是在10℃条件下加入的。
[0081]
在一些实施方案中,所述步骤a的反应温度为室温至90℃,优选地为室温至80℃,或者,优选地为30℃至90℃;更优选地,所述步骤a的反应温度为30℃至90℃,进一步优选地为30℃至60℃。
[0082]
在一些实施方案中,所述步骤a的反应可以是在氮气氛围下进行的,也可以是在没有氮气保护的情况下进行的;优选地,所述步骤a反应是在氮气氛围下进行的。
[0083]
在一些实施方案中,所述步骤a的反应溶剂是乙腈、四氢呋喃(thf)、n-甲基吡咯烷酮(nmp)、二甲基亚砜(dmso)、n,n-二甲基乙酰胺(dmac)或n,n-二甲基甲酰胺(dmf);优选地,所述步骤a的反应溶剂是乙腈、n,n-二甲基乙酰胺或n,n-二甲基甲酰胺;更优选地,所述步骤a的反应溶剂是n,n-二甲基乙酰胺或n,n-二甲基甲酰胺。
[0084]
在一些实施方案中,所述步骤a中,以式(viii)化合物的质量计,所述反应溶剂的用量为5ml/g至50ml/g,优选10ml/g至20ml/g。
[0085]
在一些实施方案中,所述步骤b的反应是在碱b的存在下进行的;其中,所述碱b为氢氧化钠或氢氧化钾等;优选地,所述碱b为氢氧化钾。
[0086]
在一些实施方案中,所述步骤b的反应是在醇类溶剂中进行的。所述醇类溶剂优选为甲醇或乙醇。
[0087]
另一方面,本发明提供了一种制备如式(ia)所示的吡咯酰胺化合物的方法,
[0088][0089]
其中,n为0、1、2、3或4,r具有本发明所述的含义。具体地,所述制备方法如本发明所描述的方法。
[0090]
一方面,本发明提供了一种制备式(i)化合物的方法,
[0091][0092]
其中,r为h、d、f、cl、br、c
1-4
烷基、c
1-4
烷氧基、c
1-4
卤代烷基或c
1-4
卤代烷氧基;
[0093]
其中,所述方法包括如下步骤:式(ii)化合物经脱保护反应得到式(i)化合物,
[0094][0095]
其中,pg1为合适的羟基保护基,包括但不限于,烷基、烯基、烷氧基烷基、烷硫基烷基、硅基烷基、苄基或取代的苄基等;优选地,pg1为苄基或取代的苄基;更优选地,pg1为苄基、对甲基苄基或对甲氧基苄基。
[0096]
在一些实施方案中,本发明所述脱保护反应是在钯碳存在条件下于氢气氛围中进行的。
[0097]
在一些实施方案中,本发明所述脱保护反应可以是在酸性条件或酸性物质存在的条件下进行的。
[0098]
在一些实施方案中,本发明所述脱保护反应是在路易斯酸存在的条件下进行的。在又一些实施方案中,所述路易斯酸为氯化锌、氯化铝或四氯化钛等。在又一些实施方案中,所述路易斯酸为氯化锌或氯化铝等。
[0099]
在另一些实施方案中,所述酸性条件是指酸存在的条件,所述酸为适用于所述脱保护反应的合适的酸,包括但不限于盐酸、乙酸、三氟乙酸或硫酸等。优选地,所述的酸为盐酸、硫酸、三氟乙酸或它们的溶液;或者,所述酸为盐酸、乙酸、三氟乙酸或它们的溶液。
[0100]
在另一些实施方案中,所述酸性条件是氯化锌、氯化铝、四氯化钛、盐酸、硫酸、乙酸或三氟乙酸存在的条件。
[0101]
在另一些实施方案中,所述酸性条件是氯化锌、盐酸、乙酸或三氟乙酸存在的条件。
[0102]
在又一些实施方案中,以式(ii)化合物的质量计,所述酸的用量为0.17ml/g至0.34ml/g。
[0103]
在又一些实施方案中,所述盐酸是指氯化氢的水溶液。具体地,所述盐酸的浓度为大于1mol/l;优选地,所述盐酸的浓度为1mol/l至12mol/l;更优选地,所述盐酸的浓度为3mol/l至12mol/l。
[0104]
在又一些实施方案中,本发明所述的酸为盐酸,优选地,以式(ii)化合物的质量计,所述盐酸的用量为0.17ml/g至0.34ml/g。
[0105]
在一些实施方案中,本发明所述脱保护反应的反应温度为50℃至75℃;优选地,所述反应温度为55℃至70℃。
[0106]
在一些实施方案中,本发明所述脱保护反应可以在常压(0.1mpa)条件下进行,也可以在大于常压的条件下进行。在另一些实施方案中,本发明所述脱保护反应的压强是0.1mpa至3.8mpa。在又一些实施方案中,本发明所述脱保护反应的压强是0.8mpa至3.8mpa。
[0107]
在一些实施方案中,本发明所述脱保护反应是在醇类溶剂中进行的,所述醇类溶剂为甲醇、乙醇、异丙醇、叔丁醇或叔戊醇。优选地,以式(ii)化合物的质量计,所述醇类溶剂的用量可以是10ml/g。
[0108]
在一些实施方案中,本发明所述脱保护反应得到的粗品经重结晶纯化可得到纯的式(i)化合物。
[0109]
在另一些实施方案中,本发明所述重结晶是在乙醇和水的混合溶剂中进行的。在又一些实施方案中,所述乙醇和水的体积比以是任意合适的比例,包括但不限于1:2至1:15。
[0110]
在另一些实施方案中,本发明所述的重结晶可以在乙酸异丙酯和甲苯的混合溶剂中进行。在又一些实施方案中,所述乙酸异丙酯和甲苯的体积比可以是任意合适的比例,包括但不限于1:2至1:15;具体地,所述乙酸异丙酯和甲苯的体积比可以是1:5。
[0111]
本发明中所述式(i)化合物的纯化可以通过多次重结晶来实现,每次重结晶的条件可以相同或不同。必要时,本发明所述式(i)化合物可以通过本领域其他常用的纯化方法来进一步去除杂质,例如,通过使用除钯硅胶和/或活性炭来去除残留的钯。所述重结晶可以是先将粗产物溶于良性溶剂中,搅拌溶解后,加入不良溶剂或者将粗产物的溶液加入到不良溶剂中,搅拌析出固体。所述溶解过程可以是在常温条件下进行,也可以是在加热升温的条件下进行。优选地,在使用良性溶剂溶解粗产物后可以先使用除钯硅胶和/或活性炭等手段去除杂质,然后再将粗产物的溶液加入到不良溶剂中以析出固体。优选地,上述重结晶中,良性溶剂可以是乙醇或乙酸异丙酯,不良溶剂可以是水或甲苯。在一些实施方案中,良性溶剂是乙醇,不良溶剂是水;优选地,所述乙醇和水的体积比可以为约1:1.5至约1:15,更优选地可以为约1:1.8至约1:15。在另一些实施方案中,良性溶剂是乙酸异丙酯,不良溶剂是甲苯;优选地,所述乙酸异丙酯和甲苯的体积比可以为约1:2至约1:15,更优选地可以为约1:4至约1:5。
[0112]
在一些实施方案中,本发明所述式(ii)化合物可以通过本发明所述的方法制备得到。
[0113]
另一方面,本发明提供了一种制备式(viii-a)化合物的方法,其包括以下步骤:式(ix)化合物在脂肪酶的作用下得到式(viii-a)化合物;
[0114][0115]
在一些实施方案中,所述脂肪酶为诺维信novozyme 435。
[0116]
在一些实施方案中,所述反应的溶剂是甲基叔丁基醚、四氢呋喃、甲苯、2-甲基四氢呋喃、丙酮、乙腈或乙酸乙酯等;优选地,所述反应的溶剂为乙腈。
[0117]
在一些实施方案中,所述反应是在丙酸乙烯酯等酰基供体的存在下进行的。
[0118]
在一些实施方案中,所述反应的反应温度为0℃至50℃,优选地为室温或5℃至40℃;更优选地,所述反应的反应温度为5℃至10℃。
[0119]
再一方面,本发明提供了如式(ii)、式(iii)或式(viii-a)所示的化合物,
[0120][0121]
其中,r具有本发明所述的含义,pg1为合适的羟基保护基,包括但不限于,烷基、烯基、烷氧基烷基、烷硫基烷基、硅基烷基、苄基或取代的苄基等;优选地,pg1为苄基或取代的苄基。
[0122]
在一些实施方案中,本发明提供了如式(ii-1)、式(ii-2)或式(iii-1)所示化合物,
[0123][0124]
本发明所述式(iii-1)所示化合物用于制备本发明所述的式(ia)或(i)所示化合物。优选地,使用式(iii-1)所示化合物制备式(ia)或(i)所示化合物的方法如本发明所述。
[0125]
本发明提供了制备吡咯酰胺化合物的新方法,其中,所述吡咯酰胺化合物包括埃沙西林酮及其类似物。同时,本发明还提供了所述方法中的重要中间体及其制备方法。本发明的制备方法原料便宜,条件温和,操作简单,安全可控,总收率高,产品纯度高,适合工业化生产。
[0126]
具体实施方法
[0127]
定义和一般术语
[0128]
本发明中“室温”或“常温”指的是温度由大约10℃至大约40℃。在一些实施方案中,“室温”指的是温度由大约20℃至大约30℃;在另一些实施方案中,“室温”指的是20℃、22.5℃、25℃、27.5℃等等。本发明中的“r.t.”是“室温”的缩写。
[0129]
本发明中“低温”指的是温度由大约0℃至大约10℃。
[0130]
在本发明的上下文中,所有在此公开了的数字均为近似值。每一个数字的数值有可能会出现1%、2%、5%、7%、8%或10%等差异。每当公开一个具有n值的数字时,任何具有n+/-1%、n+/-2%、n+/-3%、n+/-5%、n+/-7%、n+/-8%或n+/-10%值以内的数字会被明确地公开,其中“+/
‑”
是指加或减。每当公开一个数值范围中的一个下限、dl和一个上限、du时,任何处于该公开了的范围之内的数值会被明确地公开。
[0131]
本发明所述的“产物含量”或“产物比例”指的是反应完毕后,经hplc检测的反应体系中产物的含量。
[0132]
本发明所述的所有反应步骤反应到一定程度如原料消耗大约大于70%、大于80%、大于90%、大于95%、或经检测反应原料已经消耗完毕后进行后处理,如冷却、收集、提取、过滤、分离、净化处理或其组合。可以通过常规的方法如薄层层析法(tlc)、高效液相色谱法(hplc)、气相色谱法(gc)等方法检测反应程度。可以采用常规的方法对反应溶液进行后处理,例如,通过减压蒸发或常规蒸馏反应溶剂后收集粗产物,直接投入下一步反应;或直接过滤得到粗产物,直接投入下一步反应;或静置后,倾倒出上层清液得到粗产物,直接投入下一步反应;或选择适当的有机溶剂或其组合进行萃取、蒸馏、结晶、柱层析、润洗、
打浆等纯化步骤。
[0133]
本发明中的术语“大约”或“约”、“左右”是用于修饰一个上下相差10%的数值。在一些实施方案中,“大约”或“约”、“左右”用于修饰一个上下相差5%的数值。在一些实施方案中,“大约”或“约”、“左右”用于修饰一个上下相差3%或2%或1%的数值。可以理解的是,“大约”或“约”、“左右”修饰的数值误差范围是取决于其所修饰的数值的实际或合理的误差范围。
[0134]
本发明所述各步反应过程中,反应原料或其他试剂可以通过滴加的方式加入到反应体系中。所述各滴加过程以及所述的各步反应均在一定温度条件下进行,任何适合使用于各滴加过程或各反应过程的温度均包含在本发明中。另外,本领域的许多类似改动,等同替换,或等同于本发明所描述的温度及温度范围,均视为本发明的包含范围。本发明给出了各滴加过程较佳的温度或温度范围,以及各反应较佳的反应温度或反应温度范围。
[0135]
本发明所述的“溶剂1”、“溶剂2”、“溶剂a”、“溶剂b”、“碱a”、“碱b”等表述,在“溶剂”或“碱”后面使用阿拉伯数字1、2、3
……
或者字母a、b、c
……
,仅仅是为了更好的区分各个步骤中使用的溶剂或碱,所使用的阿拉伯数字或字母并无特殊含义。例如,溶剂1,包括所有适用于由式(iii)化合物与式(iv)所示化合物反应制备式(ii)化合物的反应的溶剂,包括但不限于甲苯、二氧六环、二甲基亚砜、叔丁醇、叔戊醇、二甲醚(dme)、环戊基甲醚(cpme)、n,n-二甲基乙酰胺、水或它们的任意组合。
[0136]
本发明所述的各反应步骤所使用的溶剂没有特别限制,任何在一定程度上能溶解起始原料并且不抑制反应的溶剂均包含在本发明中。另外,本领域的许多类似改动,等同替换,或等同于本发明所描述的溶剂,溶剂组合,及溶剂组合的不同比例,均视为本发明的包含范围。本发明给出了各反应步骤所使用的较佳的溶剂。
[0137]
本发明所述的各反应步骤的产物,在合适的条件下,可以通过重结晶的方式进行纯化。所使用的重结晶溶剂没有特别限制,任何在一定程度上能溶解粗产物并且在一定条件下能析出结晶的溶剂均包含在本发明中。另外,本领域的许多类似改动,等同替换,或等同于本发明所描述的溶剂,溶剂组合,及溶剂组合的不同比例,均视为本发明的包含范围。其中,所述的溶剂可以是醇类,醚类,烷烃类,卤代烃类,酯类,酮类,芳烃类,乙腈,乙酸,水,dmf或它们的组合。例如水,乙酸,甲醇,乙醇,正丙醇,异丙醇,正丁醇,异丁醇,叔丁醇,石油醚,正戊烷,正己烷,正庚烷,环己烷,dmf,n,n-二甲基乙酰胺,四氢呋喃,乙醚,异丙醚,二氧六环,甲基叔丁基醚,二甲氧乙烷,二乙二醇二甲醚,三甘醇二甲醚,二氯甲烷,1,2-二氯乙烷,氯仿,四氯化碳,乙酸乙酯,乙酸异丙酯,丙酮,丁酮,苯,甲苯,二甲苯或它们的组合。
[0138]
本发明所述的溶剂中水分的含量,没有特别的限制,即,溶剂中水分的含量不影响本发明所述反应的发生。任何在一定程度上能在本发明中使用的含有一定量的水分的溶剂,均视为本发明所述的溶剂。如溶剂中水分的含量大约小于0.05%,小于0.1%,小于0.2%,小于0.5%,小于5%,小于10%,小于25%,小于30%,或为0%。在一些实施方案中,所述溶剂的水分含量在一定范围内,更有利于反应的进行;例如,在以乙醇作为反应溶剂的步骤,使用无水乙醇,更有利反应的进行。在一些实施方案中,所述溶剂的水分含量超出一定范围,可能会影响反应的进行(例如,影响反应的收率),但并不影响反应的发生。
[0139]
本发明中“取代的”是指所述基团可以被至少一个合理的取代基取代,所述取代基包括但不限于:卤素、氘、c
1-20
烷基、c
1-20
烷氧基、c
1-20
卤代烷基或c
1-20
卤代烷氧基,优选卤
素、氘、c
1-4
烷基、c
1-4
烷氧基、c
1-4
卤代烷基或c
1-4
卤代烷氧基。例如“取代的苄基”可以为对甲基苄基、对甲氧基苄基等。
[0140]
本发明中出现的缩写及含义如下:
[0141]
缩写含义bn苄基cdin,n'-羰基二咪唑pd2(dba)3三(二亚苄基丙酮)二钯pd(dppf)cl
2.
ch2cl21,1'-双(二苯膦基)二茂铁二氯化钯二氯甲烷络合物xantphos4,5-双(二苯基膦)-9,9-二甲基氧杂蒽s-phos2-二环己基膦-2',6'-二甲氧基联苯x-phos2-二环己基膦-2',4',6'-三异丙基联苯dmedan,n'-二甲基乙二胺tmedan,n,n',n'-四甲基乙二胺cpme环戊基甲醚dme二甲醚t-amoh叔戊醇t-buoh叔丁醇dmacn,n-二甲基乙酰胺dmfn,n-二甲基甲酰胺thf四氢呋喃dmso二甲基亚砜nmpn-甲基吡咯烷酮
[0142]
一般合成及测定方法
[0143]
在本说明书中,如果在化学名称和化学结构间存在任何差异,结构是占优的。
[0144]
下面所描述的实施例,除非其他方面表明,所有的温度定为摄氏度(℃)。除非其他方面表明,各种物料、试剂购买于商品供应商,使用时都没有经过进一步纯化;部分物料可根据本领域公知的方法制备得到。
[0145]
核磁共振光谱数据通过bruker avance 400核磁共振谱仪或bruker avance iii hd 600核磁共振谱仪来测定,以cdc13,d
6-dmso,cd3od,d2o或d
6-丙酮为溶剂(以ppm为单位),用tms(0ppm)或氯仿(7.25ppm)作为参照标准。当出现多重峰的时候,将使用下面的缩写:s(singlet,单峰),d(doublet,双峰),t(triplet,三重峰),m(multiplet,多重峰),br(broadened,宽峰),dd(doublet of doublets,双二重峰),dt(doublet of triplets,双三重峰),td(triplet of doublets,三双重峰),ddd(doublet of doublet of doublets,双双二重峰),ddt(doublet of doublet of triplets,双双三重峰),dddd(doublet of doublet of doublet of doublets,双双双二重峰)。偶合常数,用赫兹(hz)表示。
[0146]
本发明各化合物的纯度和/或对映异构体过量可以按照本发明记载的色谱分析方法进行测定。本发明中的色谱条件并不作为限定,可根据化合物及其杂质、或对映异构体的分离效果进行适当的选择或调整。
[0147]
本发明实施例公开了制备如式(ia)、式(i)、式(iia)或式(ii)所示吡咯酰胺化合
物的方法。本领域技术人员可以借鉴本发明内容,适当改进工艺参数来实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明中。本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法进行改动或适当变更与组合,来实现和应用本发明技术。
[0148]
为了使本领域的技术人员更好地理解本发明,下面结合实施例对本发明进行详细说明。
实施例
[0149]
实施例1中间体(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0150][0151]
步骤1:(s)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯的制备
[0152][0153]
将1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯(3073g,9389mmol)溶于乙腈(30.7l)中,体系澄清后加入丙酸乙烯酯(1880ml,16900mmol)和诺维信435脂肪酶(92.2g),继续室温搅拌,hplc监测反应。反应3.5h后,停止反应。反应液过滤除去脂肪酶,滤液减压浓缩,残留物经硅胶柱层析分离(石油醚/乙酸乙酯(v/v)=1/1),得淡黄色透明油状物(841g,27.37%)。hplc纯度97.03%,ee值93.84%。
[0154]
目标化合物纯度使用高效液相色谱法进行测定:
[0155]
空白溶液:甲醇;供试品溶液:取供试品,用甲醇配制成浓度约为1mg/ml的溶液。流动相a:0.05%磷酸水溶液;流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=9:1~2:8)。色谱柱:xbridge phenyl(4.6
×
150mm,3.5μm);流速:1.0ml/min;检测波长:238nm;柱温:45℃;进样量:4μl;停止时间:40min。
[0156]
目标化合物的对映异构体过量使用以下方法进行测定:
[0157]
空白溶液:乙醇;供试品溶液:取供试品,用乙醇配制成浓度约为1mg/ml的溶液。流动相a:1%三氟乙酸-乙醇溶液;流动相b:正己烷;洗脱程序:等度洗脱(流动相a:流动相b=3:97)。色谱柱:chiral as-h(4.6mm
×
250mm,5μm);流速:0.7ml/min;检测波长:238nm;柱温:25℃;进样量:3μl;停止时间:50min。
[0158]
步骤2:(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯的制备
[0159][0160]
将(s)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯(1580g,4827mmol)和苄溴(1432g,8205mmol)加入dmac(31600ml)中,降温至10℃,分批加入叔戊醇钠(922.2g,8206mmol),加毕,氮气置换釜内空气并升温至40℃,搅拌4h,停止反应。反应液倒入冰水(50l)中,乙酸乙酯(5l
×
2)萃取,合并有机相,经饱和食盐水(10l
×
2)洗涤,收集有机相,减压蒸干,得褐色油状物(1960g,97.27%)。
[0161]
目标化合物纯度或产物含量使用高效液相色谱法进行测定:
[0162]
空白溶液:甲醇;供试品溶液:取供试品,用甲醇10倍稀释以得待测样品。流动相a:0.05%磷酸水溶液;流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=8:2~2:8)。色谱柱:xbridge phenyl(4.6
×
150mm,3.5μm);流速:1.0ml/min;检测波长:238nm;柱温:40℃;进样量:1μl;停止时间:40min。
[0163]
参照上述方法,以(s)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯为原料,对反应的溶剂种类及其用量、碱的种类和用量、苄溴的用量、反应温度等条件进行筛选。实验结果如表a所示。由表a可知,不同的碱或不同的溶剂对反应的影响较大。
[0164]
表a
[0165]
[0166][0167]
注:
[0168]
当量,是指以(s)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯的摩尔量为标准,其他物料的摩尔量的比例。
[0169]
示例27是在没有氮气保护的条件下进行的。
[0170]
除非另外说明,表a中溶剂的用量一般是20ml/g,即,每1g的(s)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯,使用的溶剂为20ml。
[0171]
步骤3:(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸的制备
[0172][0173]
将(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸甲酯(1960g,4696mmol)溶于甲醇(7840ml)和水(1960ml)的混合溶剂中,缓慢加入氢氧化钾(1240g,18786mmol,85mass%),升温至70℃,冷凝回流,搅拌4h,停止反应。减压蒸去溶剂,加水(8l)稀释反应液,甲基叔丁基醚(8l)和石油醚(4l)的混合溶剂洗涤,所得水相在10℃下倒入至溶有浓盐酸(2000ml,12mol/l)的水(15l)溶液中,打浆过夜,抽滤,烘干得淡黄色固体(1785g,94.23%)。hplc纯度99.38%。
[0174]
目标化合物纯度使用高效液相色谱法进行测定:
[0175]
空白溶液:甲醇;供试品溶液:取供试品,用乙醇配制成浓度约为0.8mg/ml的溶液。流动相a:0.05%磷酸水溶液;流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=8:2~1.5:8.5)。色谱柱:xbridge phenyl(4.6
×
150mm,3.5μm);流速:1.0ml/min;检测波长:220nm;柱温:30℃;进样量:7μl;停止时间:36min。
[0176]
步骤4:(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0177][0178]
方法一:
[0179]
示例1:
[0180]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸(129.5g,321.0mmol)于1000ml烧瓶中,向其中加入n,n-二甲基甲酰胺(260ml),分批加入n,n'-羰基二咪唑(74.6g,451mmol,98mass%),室温反应2h。冰浴条件下,向氨水(520ml,3460mmol,26mass%)中加入上述反应液,完毕后移至室温反应15h。向体系中加入稀盐酸(1m,520ml),室温继续反应3h。抽滤,滤饼用水洗涤(100ml
×
2),收集滤饼,60℃真空干燥12h,得粗产品为类白色固体(127g,98.31%)。
[0181]
称取粗品(10g)于100ml烧瓶中,向其中加入乙酸叔丁酯(30ml),升温至85℃,加入环己烷(30ml),于85℃下搅拌2h后关闭加热。降至室温后继续搅拌3h。抽滤,滤饼用环己烷/乙酸叔丁酯((v/v)=1/1,20ml)洗涤,收集滤饼,60℃真空干燥,得到类白色固体(9.37g,93.7%)。hplc纯度99.84%,ee值100%。
[0182]
目标化合物纯度使用高效液相色谱法进行测定:
[0183]
空白溶液:甲醇;供试品溶液:取供试品,用甲醇配制成浓度约为1.0mg/ml的溶液。流动相a:0.05%氨水-水溶液(磷酸调节ph至8);流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=7:3~2:8)。色谱柱:zorbax extend-c18(4.6
×
150mm,5μm);流速:1.0ml/min;检测波长:238nm;柱温:30℃;进样量:6μl;停止时间:25min。
[0184]
目标化合物的对映异构体过量使用以下方法进行测定:
[0185]
空白溶液:甲醇;供试品溶液:取供试品,用乙醇配制成浓度约为1mg/ml的溶液。流动相a:1%三氟乙酸-乙醇溶液;流动相b:正己烷;洗脱程序:等度洗脱(流动相a:流动相b=1:9)。色谱柱:chiralpak as-h(4.6
×
250mm,5μm);流速:1.0ml/min;检测波长:238nm;柱温:30℃;进样量:6μl;停止时间:40min。
[0186]
示例2:
[0187]
将(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸(1785g,4425mmol)溶于dmf(3570ml)中,室温下分批加入cdi(952g,5754mmol),继续室温搅拌1h,停止反应。将反应液直接用于下一步,收率以100%计。
[0188]
10℃下,将如上制备得到的(s)-(1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-基)(1h-咪唑-1-基)甲酮(2007g,4426mmol)的dmf(3570ml)反应液缓慢
加入至氨水(7000ml,26mass%)中,继续在10℃下搅拌3h后,缓慢升至室温,搅拌过夜。加入盐酸(8l,1.5mol/l),搅拌1h,抽滤,烘干,得粗产品为类白色固体(1700g,95.46%)。
[0189]
将所得粗品(1700g,4225mmol)溶于乙酸异丙酯(5100ml)中,升温至80℃左右搅拌30min至体系澄清,加入环己烷(5100ml),继续搅拌1h。关闭加热,搅拌过夜。抽滤,所得类白色固体的ee值为99.44%,hplc纯度为99.97%。
[0190]
当环己烷和乙酸异丙酯的体积为2:1时,或者,用乙酸叔丁酯替换乙酸异丙酯,同样可以实现纯化效果。例如:用乙酸叔丁酯替换乙酸异丙酯进行重结晶纯化(环己烷/乙酸叔丁酯(v/v)=3/4,用量为每克粗产品7ml混合溶剂),所得产品的hplc纯度为99.94%,ee值为99.78%。
[0191]
方法二:
[0192]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸(10.0g,24.8mmol)于100毫升烧瓶中,向其中加入n,n-二甲基甲酰胺(20ml)和n,n'-羰基二咪唑(4.92g,29.76mmol),完毕后室温反应2h。向体系中加入硫氰酸铵(7.65g,99.0mmol)和碳酸钾(12.8g,91.7mmol),室温继续反应12h。向体系中加入水(80ml),室温继续搅拌2h。抽滤,滤饼用水洗涤(20ml
×
2),收集滤饼,60℃真空干燥12h,得到类白色固体(9.50g,95.2%),hplc纯度99.21%,ee值99.02%。
[0193]
方法三:
[0194]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸(10.0g,24.8mmol)于100ml反应瓶中,向其中加入n,n'-羰基二咪唑(4.92g,29.76mmol)和n,n-二甲基甲酰胺(20ml),完毕后室温反应2h。向体系中加入溴化铵(12.2g,123mmol)和碳酸钾(12.8g,91.7mmol),室温继续反应17h。向体系中加入水(80ml),室温搅拌30min。抽滤,滤饼用水洗涤(10ml
×
3),收集滤饼,60℃真空干燥12h,得到类白色固体(9.40g,94.6%),hplc纯度98.92%,ee值98.64%;ms(esi,pos.ion)m/z:403.2(m+1)。
[0195]
参照上述方法二和方法三,以不同的氨基化试剂或不同用量的氨基化试剂进行产品制备,结果如表b所示。由表b可知,不同的氨基化试剂对反应的进行影响大,例如,同样条件下,以氯化铵或碳酸氢铵为氨基化试剂制备目标酰胺化合物,产率低。
[0196]
表b
[0197]
示例氨基化试剂用量产率1nh4cl5当量40%2nh4br3当量93.5%3nh4hco35当量40%4nh
3.
h2o10当量98.3%
[0198]
注:当量,是指以(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸的摩尔量为计,其他物料的摩尔量的比例。
[0199]
方法四:
[0200]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酸(10g,24.79mmol)于250毫升烧瓶中,向其中加入二氯甲烷(100ml)和3滴n,n-二甲基甲酰胺,冰浴条件下滴加草酰氯(3.2ml,38mmol),完毕后室温反应2h。减压旋蒸除去溶剂,再加入四氢呋喃(50ml)使其全部溶解。冰浴条件下将上述溶液滴加至氨水(100ml,1330mmol,
25mass%)中,完毕后移至室温反应10h。向体系中加入水(100ml),室温继续搅拌3h。抽滤,滤饼用水(20ml)洗涤,收集滤饼,60℃真空干燥12h,得淡黄色粗产物。
[0201]
向粗产物中加入乙酸异丙酯(28.5ml),升温至80℃滴加环己烷(57ml),继续在80℃下搅拌1h后关闭加热,降至室温后继续搅拌15h。抽滤,滤饼用环己烷/乙酸异丙酯((v/v)=2/1,28ml)洗涤,收集滤饼,60℃真空干燥12h,得到类白色固体(9.39g,94%),hplc纯度99.75%,ee值100%;ms(esi,pos.ion)m/z:403.2(m+1)。
[0202]
实施例2(s)-n-(3-氟-4-(甲基磺酰基)苯基)-1-(2-羟乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0203]
步骤1:(s)-1-(2-(苄基氧基)乙基)-n-(3-氟-4-(甲基磺酰基)苯基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0204][0205]
向(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(2.12kg,5.268mol)中加入叔戊醇(13.8kg)、4-溴-2-氟-1-(甲基磺酰基)苯(1.38kg,5.453mol)、碳酸铯(3.45kg,10.59mol)、醋酸钯(31.8g,141.6mmol)和4,5-双二苯基膦-9,9-二甲基氧杂蒽(xantphos)(77.0g,133.0mmol),反应体系置换3次氮气,然后加热至65℃反应4h。终止反应后向体系中加入水(10.90kg),搅拌5min后再加入乙酸异丙酯(5.93kg)稀释反应液,搅拌5min后静置分液。分出下层水相,上层有机相用硅藻土抽滤,滤饼用乙酸异丙酯(5.89kg)洗涤,滤液减压浓缩,向浓缩物中加入乙醇(9.95kg),10℃条件下,向水(91.20kg)中滴加上述混合液;滴加完毕继续搅拌5h。抽滤,滤饼用水(4.85kg)洗涤,收集滤饼,45℃下真空干燥48h,得粗产品。
[0206]
向粗产品中加入乙醇(9.30kg),使其全部溶解后再加入除钯硅胶(470g)和活性炭(170g),然后加热至70℃,搅拌1h后关闭加热,降至室温后继续搅拌12h。硅藻土过滤,滤饼用乙醇(4.65kg)洗涤,收集滤液。10℃条件下,向水(91.55kg)中滴加上述滤液,滴加完毕继续搅拌5h。抽滤,滤饼用水(5.50kg)洗涤,收集滤饼,45℃下真空干燥48h,得类白色固体(2.95kg,97.4%),hplc纯度95.0%,ee值98.8%,pd含量55.68ppm。
[0207]
目标化合物纯度使用高效液相色谱法进行测定:
[0208]
空白溶液:甲醇;供试品溶液:取供试品,用乙醇配制成浓度约为1.0mg/ml的溶液。流动相a:1.36g/l磷酸二氢钾水溶液(磷酸调节ph至2.0);流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=6.5:3.5~2:8)。色谱柱:xbridge phenyl(4.6
×
150mm,3.5μm);流速:1.0ml/min;检测波长:210nm;柱温:35℃;进样量:4μl;停止时间:35min。
[0209]
目标化合物的对映异构体过量使用以下方法进行测定:
[0210]
空白溶液:乙醇;供试品溶液:取供试品,用乙醇配制成浓度约为2mg/ml的溶液。流动相a:1%三氟乙酸-乙醇溶液;流动相b:正己烷;洗脱程序:等度洗脱(流动相a:流动相b=3:7)。色谱柱:chiral ny(4.6
×
250mm,5μm);流速:1.0ml/min;检测波长:290nm;柱温:30℃;进样量:6μl;停止时间:40min。
[0211]
(1)参照上述操作对反应条件如催化剂、配体、碱、溶剂和反应温度等进行筛选,实验结果如下表c所示。由表c可知,该范围在以cui作催化剂、使用常用配体如8-羟基喹哪啶、dmeda或tmeda时,反应基本不进行或反应效果极差;而使用本发明所述钯催化剂、结合适当的配体,反应效果较佳。此外,碱对反应有影响,例如,使用cf3coona,反应效果差。
[0212]
表c
[0213]
(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(1.00kg,1.740mol)中加入乙醇(8.45kg)、钯碳(104g,10mass%)和盐酸溶液(167g,6mol/l),完毕后于氢气气氛下(氢气袋)加热至55℃反应6h。终止反应后,反应液经硅藻土抽滤,滤饼用乙醇(1.61kg)洗涤,滤液减压蒸去溶剂,往残留物中加入乙醇(2.50kg)使其全部溶解。10℃条件下,向水(22.50kg)中滴加上述混合溶液,滴加完毕后搅拌3h。抽滤,滤饼用水(2.10kg)洗涤,收集滤饼,60℃真空干燥24h,得粗产品。
[0226]
往粗产品中加入乙酸异丙酯(2.25kg)和甲苯(1.50kg),加热至55℃,再加入甲苯(8.70kg),55℃下搅拌1h后关闭加热,降至室温后继续搅拌7h。抽滤,滤饼用甲苯(1.50kg)洗涤,收集滤饼,60℃真空干燥8h。往干燥后的滤饼中加入乙醇(1.90kg),除钯硅胶(75g)和活性炭(40g)。完毕后升温至60℃搅拌1h,降至室温并继续搅拌10h。抽滤,滤饼用乙醇(1.30kg)洗涤,收集滤液。室温条件下,向上述滤液中滴加水(3.00kg),搅拌2h后再次滴加水(4.50kg),室温继续搅拌5h。抽滤,滤饼用水(1.70kg)洗涤,收集滤饼,60℃真空干燥15h,得白色固体(747.2g,88.6%),hplc纯度99.66%,ee值100%,pd含量《1ppm,水分含量为0.07%。
[0227]1h nmr(400mhz,dmso-d6)δ(ppm):10.15(s,1h),7.99(dd,j=13.5,1.4hz,1h),7.89(d,j=7.7hz,1h),7.82(s,1h),7.78(d,j=8.3hz,2h),7.75-7.67(m,2h),7.48(d,j=7.4hz,1h),4.95(t,j=5.0hz,1h),3.70(ddd,j=15.2,8.6,4.6hz,1h),3.58-3.44(m,3h),3.28(s,3h),1.93(s,3h)。
[0228]
目标化合物纯度使用高效液相色谱法进行测定:
[0229]
空白溶液:甲醇;供试品溶液:取供试品,用乙醇配制成浓度约为0.8mg/ml的溶液。流动相a:0.1%磷酸水溶液;流动相b:乙腈;洗脱程序:梯度洗脱(流动相a:流动相b=7.5:2.5~1:9)。色谱柱:waters xbridge phenyl(4.6
×
150mm,3.5μm);流速:1.0ml/min;检测波长:215nm;柱温:40℃;进样量:5μl;停止时间:38min。
[0230]
目标化合物的对映异构体过量使用以下方法进行测定:
[0231]
空白溶液:乙醇;供试品溶液:取供试品,用乙醇配制成浓度约为1.2mg/ml的溶液。流动相a:1%三氟乙酸-乙醇溶液;流动相b:正己烷;洗脱程序:等度洗脱(流动相a:流动相b=1.5:8.5)。色谱柱:chiralpak ic(4.6
×
250mm,5μm);流速:1.0ml/min;检测波长:290nm;柱温:25℃;进样量:8μl;停止时间:65min。
[0232]
(1)参照上述方法操作,对酸或酸性物质进行筛选试验,结果如下表e所示。
[0233]
表e
[0234]
示例氢气压力酸或酸性物质产物含量hplc纯度ee值1常压(氢气袋)三氟乙酸93.54%99.85%99.91%2常压(氢气袋)氯化铝92.23%99.84%99.95%3常压(氢气袋)四氯化钛93.00%99.80%99.95%4常压(氢气袋)6m h2so492.31%99.80%99.95%
[0235]
(2)参照上述方法操作,对盐酸溶液的浓度和用量、氢气压强、反应温度等条件进行筛选试验,结果如下表f所示。
[0236]
表f
[0237][0238]
实施例3(s)-1-(2-(羟乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0239]
步骤1:(s)-1-(2-(苄氧基)乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0240][0241]
示例1:
[0242]
氮气保护下,将(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(50.0g,124mmol)、1-碘-4-(甲磺酰基)苯(35.1g,124mmol)、醋酸钯(1.39g,6.19mmol)、xantphos(3.59g,6.20mmol)和碳酸铯(80.8g,249mmol)加入到叔戊醇(500ml)中,升温至80℃,搅拌6h,停止反应。反应液经硅藻土过滤,滤液加水(500ml)稀释,乙酸乙酯(200ml
×
2)萃取,合并有机相,经饱和食盐水(200ml
×
2)洗涤,收集有机相,减压蒸去溶剂,残留物经(乙醇/水(v/v)=100ml/1500ml)重结晶,抽滤,得类白色固体(60.0g,86.8%)。ms(esi,pos.ion)m/z:557.5(m+1)。
[0243]
示例2:
[0244]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰
胺(300.0g,745.5mmol)于10l反应瓶中,向其中加入4-溴-1-(甲基磺酰基)苯(192.8g,820.1mmol)、碳酸铯(485.8g,1461mmol)、叔戊醇(2400ml)、醋酸钯(5.02g,22.4mmol)和4,5-双二苯基膦-9,9-二甲基氧杂蒽(12.9g,22.3mmol),反应体系置换3次氮气,然后加热至65℃加热反应4h。终止反应后向体系中加入水(1500ml),搅拌5min后再加入乙酸异丙酯(900ml)稀释反应液,搅拌5min后静置分液。分出下层水相,上层有机相用硅藻土抽滤,滤饼用乙酸异丙酯(600ml)洗涤,滤液减压浓缩。向浓缩物中加入乙醇(1660ml),使其全部溶解后再加入除钯硅胶(40g)和活性炭(40g)。加热至70℃搅拌1h后关闭加热,降至室温后继续搅拌12h。硅藻土过滤,滤饼用乙醇(830ml)洗涤,收集滤液。冰浴条件下,向水(7000ml)中滴加上述滤液。滴加完毕,移至室温并继续搅拌5h。抽滤,滤饼用水(700ml)洗涤,收集滤饼,45℃下真空干燥48h,得类白色固体(401.35g,96.72%)。ms(esi,pos.ion)m/z:557.2(m+1);hplc纯度99.13%,ee值98.28%,pd含量7.90ppm。
[0245]
目标化合物的纯度、其对映异构体过量的测定方法可参照实施例2步骤1的测定方法。
[0246]
将上述方法中的xantphos配体替换为x-phos或s-phos、醋酸钯替换为pd2(dba)3或pd(dppf)cl2·
ch2cl2、碳酸铯替换为磷酸钾、叔丁醇钾或碳酸钾,参照上述操作步骤同样可以制备得到目标化合物。部分实验结果如下表g所示。
[0247]
表g
[0248][0249]
注:除非另外说明,上述表g各示例中,以(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的摩尔量为基础,溴底物4-溴-1-(甲基磺酰基)苯的
用量是1.1当量,碱的用量是2.0当量,催化剂和配体的用量以百分摩尔比计算,具体用量如上表所示;以(s)-1-(2-(苄氧基)乙基)-4-甲基-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的质量为基础,溶剂的用量为10ml/g。
[0250]
步骤2:(s)-1-(2-(羟乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺的制备
[0251][0252]
示例1:
[0253]
将(s)-1-(2-(苄氧基)乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(50.0g,89.8mmol)、盐酸(7.5ml,6mol/l)和钯碳(5.0g,10%)加入到乙醇(1000ml)中,氢气置换高压釜内空气并充至2mpa,升温至50℃搅拌10h,停止反应。反应液经硅藻土过滤,滤液减压蒸干,粗产品经(乙醇/水(v/v)=100ml/1.5l)重结晶,抽滤,得白色固体(38.0g,90.7%)。
[0254]
hplc纯度:99.32%。
[0255]
ms(esi,pos.ion)m/z:467.4(m+1)。
[0256]1h nmr(400mhz,cdcl3)δ(ppm)7.95
–
7.87(m,3h),7.86
–
7.79(m,3h),7.69
–
7.58(m,2h),7.52(s,1h),7.38(d,j=7.3hz,1h),3.82
–
3.64(m,4h),3.07(s,3h),2.10(s,3h)。
[0257]
示例2:
[0258]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(10.0g,24.9mmol)于250毫升烧瓶中,向其中加入4-碘-1-(甲基磺酰基)苯(7.71g,27.3mmol)、碳酸铯(16.5g,49.6mmol)、叔戊醇(80ml)、醋酸钯(168mg,0.748306mmol)和4,5-双二苯基膦-9,9-二甲基氧杂蒽(431mg,0.7449mmol),反应体系置换3次氮气,完毕后加热至65℃反应4h。关闭加热,向体系中加入水(50ml),搅拌3min使体系中的无机盐全部溶解,再加入乙酸异丙酯(30ml)稀释反应液,搅拌3min后静置分液。分出下层水相,上层有机相用硅藻土抽滤,滤饼用乙酸异丙酯(20ml)洗涤,滤液减压浓缩得粗产品。
[0259]
向粗产品中加入乙醇(55ml),使其全部溶解后再加入除钯硅胶(13g)和活性炭(13g),完毕后加热至70℃搅拌1h后关闭加热,降至室温后继续搅拌10h。硅藻土过滤,滤饼用乙醇(27ml)洗涤,收集滤液。冰浴条件下,向水(210ml)中滴加上述乙醇滤液。滴加完毕,移至室温并继续搅拌10h。抽滤,滤饼用水(25ml)洗涤,抽滤,收集滤饼,45℃下真空干燥24h,得类白色固体(13.3g,96.2%)。
[0260]
示例3:
[0261]
称取(s)-1-(2-(苄氧基)乙基)-4-甲基-n-(4-(甲基磺酰基)苯基)-5-(2-(三氟甲基)苯基)-1h-吡咯-3-甲酰胺(200.0g,359.3mmol)于5000ml四口烧瓶中,向其中加入乙醇(2000ml)、钯碳(20g,10mass%)和盐酸溶液(24ml,144mmol,6mol/l),然后在氢气气氛下(氢气袋)加热至55℃反应6h,hplc监控。终止反应,反应液经硅藻土抽滤,滤饼用乙醇
(400ml)洗涤,滤液减压蒸去溶剂,往残留物中加入乙醇(500ml)使其全部溶解,冰浴条件下,向水(3340ml)中滴加上述溶液。滴加完毕后移至室温搅拌8h后抽滤,滤饼用水(600ml)洗涤,收集滤饼,60℃真空干燥16h。往干燥后的固体中加入乙酸异丙酯(500ml),加热至55℃,再加入甲苯(2000ml),55℃搅拌1h后关闭加热,自然降至室温并继续搅拌7h后抽滤,滤饼用甲苯(400ml)洗涤,收集滤饼,60℃真空干燥12h得粗产品。
[0262]
往粗产品中加入乙醇(668ml),除钯硅胶(16.8g)和活性炭(8.4g)。完毕后升温至60℃加热搅拌1h后关闭加热,自然降至室温并继续搅拌17h后抽滤,滤饼用乙醇(167ml)洗涤,收集滤液。室温条件下,向上述滤液中滴加水(1670ml)。室温搅拌9h后抽滤,滤饼用水(600ml)洗涤,收集滤饼,60℃真空干燥15h,得白色固体(149.6g,89.25%),hplc纯度99.84%,ee值100%,pd含量《1ppm。ms(esi,pos.ion)m/z:467.10(m+1)。
[0263]
目标化合物的纯度、其对映异构体过量的测定方法可参照实施例2步骤2的测定方法。
[0264]
参照上述方法,将6mol/l的盐酸溶液替换为三氟乙酸、硫酸、冰乙酸或zncl2等(用量为0.4-0.5当量),或者,将常压氢气袋换为高压釜(如2.8mpa等),同样可以制备得到目标化合物。部分实验结果如下表h所示。
[0265]
表h
[0266]
示例氢气压力酸或酸性物质温度时间产物含量12.8mpan/a60℃14h98.42%22.8mpa6m hcl60℃4h98.47%32.8mpa冰乙酸60℃4h98.35%4常压(氢气袋)6m hcl60℃12h94.49%5常压(氢气袋)冰乙酸60℃12h98.24%6常压(氢气袋)6m hcl50℃8h97.21%7常压(氢气袋)6m hcl55℃6h98.74%8常压(氢气袋)三氟乙酸55℃6h98.72%9常压(氢气袋)n/a55℃6h95.61%10常压(氢气袋)3m hcl55℃6h96.69%11常压(氢气袋)1m hcl55℃6h93.30%12常压(氢气袋)zncl255℃6h96.31%13常压(氢气袋)浓盐酸55℃6h97.29%14常压(氢气袋)6m h2so455℃6h95.23%15常压(氢气袋)氯化铝55℃6h97.60%16常压(氢气袋)四氯化钛55℃6h97.43%
[0267]
注:n/a表示没有添加该试剂或该物质,或该试剂或该物质不存在。
[0268]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。
[0269]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例
性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1