一种抗冠状病毒多肽、药物组合物及其应用
1.本发明涉及医药技术领域,具体为一种抗冠状病毒多肽、药物组合物及其应用,其具有抑制新型冠状病毒的活性,可用于制备相关抗冠状病毒药物。
背景技术:
2.新型冠状病毒(sars-cov-2)是一种传染性和致病性极强的病毒,由它导致的新冠肺炎疫情对全球产生了不可估量的损失。
3.寻找能够抑制新型冠状病毒的手段非常重要。目前,主要有疫苗、小分子药物以及多肽药物三种抗病毒药物。而在这其中,多肽以其亲和力高、选择性好、毒性低等优点在药物开发中占据着重要地位。
4.冠状病毒中的s蛋白在细胞毒力和病理发生上起着至关重要的作用,s蛋白由s1和s2两个亚基组成。其中s2中的七肽重复序列hr1和hr2能够形成六螺旋束结构的6-hb,这种结构有利于细胞膜和病毒膜的融合,从而有利于病毒的入侵。而当多肽融合抑制剂与s2中的hr1相结合后,hr1就不能与hr2 结合生成6-hb,从而达到抑制病毒入侵的效果。
5.依据这个理论,姜世博课题组从oc43冠状病毒hr2中截取出oc43-hr2p,发现它具有广谱抗病毒效果,并对其进行修饰,得到了广谱抗病毒多肽ek1 (sldqinvtfldleyemkkleeaikkleesyidlkel-nh2)。
6.尽管多肽具有亲和力高、选择性好、毒性低等优点,具有半衰期短、构象不稳定、易被酶水解、透膜性差等的缺点。因此,多个课题组开发了订书肽的策略来提高成药性。订书肽是一种稳定性高、透膜性好的多肽模拟物。通过在i,i+4或i,i+7的位置上引入不同长度侧链带有α-甲基的非天然氨基酸,再经过一个复分解反应后合成订书肽,通过这种方式合成的订书肽的稳定性、螺旋度和细胞通透性都有很大的提升。
7.因为丙氨酸的体积小、且二级结构中的结构能高,更有利于稳定二级结构,因此,我们结合订书肽以及丙氨酸替换的优势,设计并合成一系列基于 ek1的衍生物,以期获得更好的抗病毒活性。
技术实现要素:
8.(一)解决的技术问题
9.针对现有技术的不足,本发明提供了ek1及其衍生物及的制备方法与应用,利用订书肽策略并替换相应残基,使衍生物具备较好的抗病毒活性等优点,并希望提高衍生物的稳定性以及透膜性。
10.(二)技术方案
11.为实现上述增强其细胞渗透性、提高酶稳定性和抗病毒性的目的,本发明提供如下技术方案:
12.一种抗冠状病毒多肽,所述多肽为下列式1-4所示结构的多肽类活性分子中的一种,结构式如图1-4所示:
13.1.以sldqinvtfldleyemkkleeaikkleesyidlkel-nh2为肽链模板,其中21 位的e被s5和17k被s5替换并环合,并将10号位的l替换成a。
14.2.以sldqinvtfldleyemkkleeaikkleesyidlkel-nh2为肽链模板,其中 21位的e被s5和17k被s5替换并环合,并将12号位的l替换成a。
15.3.以sldqinvtfldleyemkkleeaikkleesyidlkel-nh2为肽链模板,其中 21位的e被s5和17k被s5替换并环合。并将14号位的y替换成a。
16.4.以sldqinvtfldleyemkkleeaikkleesyidlkel-nh2为肽链模板,其中 21位的e被s5和17k被s5替换并环合。并将19号位的l替换成a。
17.其中,s5表示(2r)-2-氨基-2-甲基-6-庚烯酸,片段中成对的(2r)-2
‑ꢀ
氨基-2-甲基-6-庚烯酸通过烯烃复分解反应进行环合。
18.在本文中,“本发明的多肽类活性分子”指的是本发明中具有式1-4所示结构多肽,在本文中,这种多肽可以称为“多肽片段”或“本发明多肽”。
19.式1-4中多肽的n末端的氨基和c末端的羧基以及氨基酸侧链基团可以不进行修饰,也可以在基本不影响本发明多肽活性的前提下进行修饰,如形成“药学上可接受的酯”,n末端氨基基团的修饰包括但不限于脱氨基、n-低级烷基、n-二低级烷基和n-酰基修饰,c末端羧基基团的修饰包括但不限于酰胺、低级烷基酰胺、二烷基酰胺和低级烷基酯修饰,本发明多肽c末端的羧基进行酰胺化修饰,即是-nh2。
20.本文所使用的多肽及氨基酸和化学基团的表示方法均为所属领域公认的表示方法,其中氨基酸的缩写可参照表1中的定义。特殊氨基酸结构可参照表2中的定义,在本文中,若不特别指出,氨基酸一般指l-型的氨基酸。
21.表1氨基酸缩写表
22.氨基酸三字母缩写一字母缩写氨基酸三字母缩写一字母缩写丙氨酸alaa亮氨酸leul精氨酸argr赖氨酸lysk天冬酰胺asnn蛋氨酸metm天冬氨酸aspd苯丙氨酸phef半胱酰胺cysc脯氨酸prop谷氨酰胺glnq丝氨酸sers谷氨酸glue苏氨酸thrt甘氨酸glyg色氨酸trpw组氨酸hish酪氨酸tyry异亮氨酸ilei缬氨酸valv
23.表2特殊氨基酸缩写表
24.氨基酸缩写(2r)-2-氨基-2-甲基-6-庚烯酸s5(2r)-2-氨基-2-甲基-9-癸烯酸r8[0025]“药物上可接受的盐”指一些小分子酸性或碱性化合物与多肽形成的盐,一般能够增加多肽的溶解性,所形成的盐基本上不改变多肽的活性。
[0026]
例如,通常能与本发明多肽形成盐的酸有盐酸、磷酸、硫酸、乙酸、琥珀酸、马来酸
和柠檬酸等;能与本发明多肽形成盐的碱有碱金属或碱土金属的氢氧化物、铵和碳酸盐等。
[0027]
本发明多肽的抗病毒作用可以通过所属领域常规的实验方法来验证,如细胞学实验等,在本发明的具体实施方式中,优选假病毒中和试验检测多肽的初步抗病毒效果。通过该试验,发现本发明所涉及的式1-4多肽活性分子都具有体外抗假病毒作用。
[0028]
此外,本发明要解决的另一技术问题是提供了含有式1-4结构的多肽类活性分子的药物组合物,其可以用于治疗或制备抗新冠病毒药物。
[0029]
该组合物可以含有本发明的环肽类活性分子中的一种或多种,优选仅含一种。
[0030]
该组合物可以含有一种或多种药学上可接受的稀释剂、赋形剂或载体,优选该组合物为单位剂量形式,如片剂、膜剂、丸剂、胶囊(包括持续释放或延迟释放形式)、粉剂、颗粒剂、糖浆剂或乳液剂、消毒的注射用溶液、悬浮液或冻干粉末针剂、气雾剂或液体喷剂、滴剂自动注射装置或栓剂。
[0031]
上述活性药物组分可以与一种无毒的药物学可接受的惰性载体组合在一起,如乙醇、甘油、水或其组合,本发明式(i)的多肽类活性分子优选使用消毒的注射用水溶液。
[0032]
本发明的药物组合物可通过所属领域技术人员所熟知的给药方式来进行给药,例如口服、直肠、舌下、肺部、透皮、离子透入、阴道及鼻内给药。本发明的药物组合物优选胃肠道外给药,如皮下、肌内或静脉内注射。
[0033]
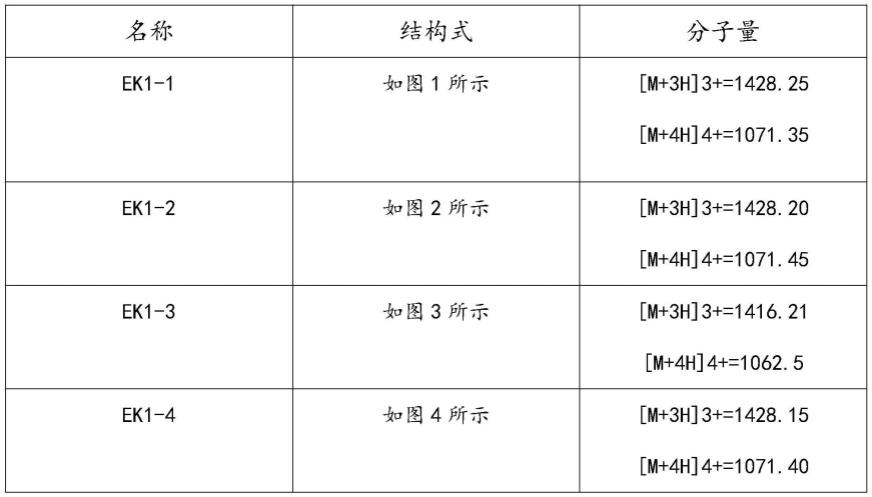
本发明合成的部分优选化合物的名称、结构式和质谱数据如表3所示
[0034]
表3优选多肽类活性分子的名称、结构式和质谱数据
[0035][0036]
为了便于理解,以下将通过具体的实施例和附图对本发明进行描述,需要特别指出的是,这些描述仅仅是示例性的描述,并不构成对本发明范围的限制。
附图说明
[0037]
图1为本发明表3中ek1-1结构式示意图;
[0038]
图2为本发明表3中ek1-2结构式示意图;
[0039]
图3为本发明表3中ek1-3结构式示意图;
cell-cell fusion%=1
‑ꢀ
(a-b)/(a0-b)
×
100%计算细胞融合抑制率
[0059]
实验结果:假病毒活性如表4所示,在假病毒感染实验中,在7.5μm浓度下几条ek1的衍生物都展现出了较好的抑制活性,通过图5可以看到,ek1-1 相比ek1表现出更好的抑制sars-cov-2假病毒效果。并且ek1-1的ic50为 34.08μm,而ek1的ic50为46.61μm,这些结果都表明经过订书肽以及丙氨酸修饰的多肽ek1-1具有更好的抑制sars-cov-2假病毒的效果。
[0060]
表4 ek1衍生物对sars-cov-2假病毒的抑制活性
[0061][0062][0063]
本发明的有益效果是:该多肽类抗病毒活性化合物及其制备方法与应用,通过订书肽策略是稳定α-螺旋肽的一种方法。这是由verdine发明的一种具有全碳氢支架的α-螺旋肽,能够增强多肽的α-螺旋结构、透膜性、抗蛋白酶水解能力、抗蛋白-蛋白相互作用的能力等。此外,丙氨酸由于其体积小、且二级结构中的结构能高,更有利于稳定二级结构,我们同时采用丙氨酸修饰,合成一系列基于ek1的衍生物。结果显示,衍生物具有一定的抗新冠病毒活性,为目前研发抗新冠病毒药物提供了新的方法。
[0064]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1