基于罗伊氏乳杆菌的CRISPR多基因编辑方法
基于罗伊氏乳杆菌的crispr多基因编辑方法
技术领域
1.本发明涉及基因工程技术领域,特别涉及基于罗伊氏乳杆菌的crispr多基因编辑方法,具体的本发明通过利用crispr系统,构建基于罗伊氏乳杆菌的多基因编辑质粒、将质粒电转化到罗伊氏乳杆菌的感受态细胞中的应用技术,提高了罗伊氏乳杆菌基因编辑效率,在技术上首次实现了对罗伊氏乳杆菌的无痕基因整合,特别适用于罗伊氏乳杆菌的连续基因编辑。
背景技术:
2.罗伊氏乳杆菌是异源发酵乳酸菌,属于革兰氏阳性兼性厌氧菌,在显微镜下呈末端圆形的略微不规则的弯曲杆菌,被发现广泛存在于人类和其他动物的肠道中。罗伊氏乳杆菌自1962年首次被分离出来,就受到广泛的关注,因为它是为数不多的能够在人体的肠胃中生存的细菌,耐受胃酸和胆盐,已被证明是人类胃肠道的常驻菌。罗伊氏乳杆菌主要定植在小肠上部的粘膜上,是新生儿及健康成年人肠道微生物菌群的主要成员。
3.罗伊氏乳杆菌对宿主健康的影响被广泛研究,主要是通过在宿主的胃肠道中定植,代谢生成一些抗菌物质和短链脂肪酸来发挥其益生功能。罗伊氏乳杆菌通过参与色氨酸代谢通路,增加肠道中吲哚衍生物及短链脂肪酸的含量,在宿主肠道中发挥免疫调节的功能;罗伊氏乳杆代谢产生的罗伊氏菌素具有广谱抗菌的能力,对肠道许多致病菌有明显的抑制作用;另外,其代谢合成的罗伊氏素还可以改变肠道肿瘤细胞附近的氧化还原平衡,抑制肿瘤的生长。
4.目前对罗伊氏乳杆菌的研究及应用多为菌体及其代谢产物的直接利用,关于罗伊氏乳杆菌的遗传体系的研究很少,国内外目前对罗伊氏乳杆菌的遗传操作是通过传统的同源重组来进行遗传改造,随着研究的不断深入,研究人员不断对其遗传操作的技术进行优化,比如利用rect介导高效的同源重组,或者是利用crispr-cas9作为反筛工具来高效的筛选得到遗传改造过的菌株,但是只能在罗伊氏乳杆菌中只能进行基因的敲除和点突变,无法无痕的整合基因,因此很大程度的限制了对罗伊氏乳杆菌相关益生机制的研究及对该菌的益生性能的应用。
技术实现要素:
5.本发明的目的是提供基于罗伊氏乳杆菌的crispr多基因编辑方法,以解决上述现有技术存在的问题,通过利用罗伊氏乳杆菌的crispr系统,一次就可以实现多个基因的编辑,为获得在宿主肠道中益生性能更优良的目的菌株奠定基础。
6.为实现上述目的,本发明提供了如下方案:
7.本发明提供一种基于罗伊氏乳杆菌的基因编辑质粒,包括:两个或者两个以上的重复序列、含有两个三型内切酶酶切位点的mini-crispr序列以及罗伊氏乳杆菌的穿梭质粒;
8.其中,每相邻的两个所述重复序列间插入所述mini-crispr序列,之后再与所述穿
梭质粒连接,即得基于罗伊氏乳杆菌的基因编辑质粒。
9.优选的是,所述重复序列如seq id no:1所示。
10.seq id no:1如下所示:gttttagatgtacttcaaatcaataatgtttagaac。
11.优选的是,所述mini-crispr序列如seq id no:2所示。
12.seq id no:2如下所示:
13.gttttagatgtacttcaaatcaataatgtttagaacagagaccgacgtcggtctcagttttagatgtacttcaaatcaataatgtttagaac。
14.本发明还提供所述的基因编辑质粒的构建方法,包括以下步骤:
15.(1)在所述重复序列间插入所述mini-crispr序列后,将其与所述穿梭质粒分别进行酶切,酶切后,进行酶连接;
16.(2)酶连接后转入大肠杆菌培养,经筛选,得到阳性转化子;
17.(3)活化所述阳性转化子,进行质粒抽提,即得基因编辑质粒。
18.优选的是,步骤(1)中,所述酶切的位点为sac1、xba1;
19.步骤(2)中,所述筛选条件为含有氯霉素抗性的lb固体培养基,37℃过夜培养,挑取阳性转化子。
20.本发明还提供一种基于所述的基因编辑质粒对罗伊氏乳杆菌进行多基因敲除的方法,包括以下步骤:
21.(1)根据罗伊氏乳杆菌胞内crispr系统的pam为3’端ngg设计所要敲除的n个基因的spacer,并根据所要敲除的n个基因的上下游序列分别设计同源臂,将同源臂克隆至所述基因编辑质粒,构建n个基因的敲除质粒;
22.(2)利用电转化,将n个基因的敲除质粒转入罗伊氏乳杆菌感受态细胞中,经筛选,挑取单菌落并进行pcr验证,得到n个基因的敲除突变株;其中,n为自然数,且n≥1。
23.本发明还提供一种基于所述的基因编辑质粒对罗伊氏乳杆菌进行基因插入的方法,包括以下步骤:
24.(1)根据罗伊氏乳杆菌胞内crispr系统的pam为3’端ngg以及拟插入位点设计spacer,并根据所述拟插入位点的上下游设计同源臂,将两个所述同源臂和拟插入基因克隆至所述基因编辑质粒,构建插入质粒;
25.(2)将所述插入质粒电转至罗伊氏乳杆菌感受态细胞,经筛选,挑取单菌落并进行pcr验证,得到基因插入突变株。
26.本发明还提供一种基于所述的基因编辑质粒对罗伊氏乳杆菌进行基因点突变的方法,包括以下步骤:
27.(1)根据罗伊氏乳杆菌胞内的crispr系统的pam为3’端ngg及拟突变位点设计spacer,并根据所述拟突变位点的上下游序列设计同源臂,突变位点设计在扩增的同源臂的引物上,将两个所述带有突变位点的同源臂克隆至所述基因编辑质粒,构建突变质粒;
28.(2)将所述突变质粒电转至罗伊氏乳杆菌感受态细胞,经筛选,挑取单菌落并进行pcr验证,得到点突变菌株。
29.优选的是,还包括对含有编辑质粒却未成功发生基因编辑的罗伊氏杆菌进行处理,以获得成功发生基因编辑菌株的步骤;所述处理方法为:经过pcr验证未发生编辑的罗伊氏乳杆菌,通过在氯霉素抗性筛选条件下,传代1-2代,即可获得成功发生编辑的罗伊氏
乳杆菌。
30.本发明还提供一种利用所述的基因编辑质粒对完成基因编辑的罗伊氏乳杆菌的反筛方法,将按照所述的方法完成的基因编辑的罗伊氏乳杆菌基因突变株,在不含抗生素的mrs固体培养基上划线培养,经过1-2代传代培养,挑取单菌落进行pcr验证,筛选得到质粒丢失的目的菌株。
31.本发明公开了以下技术效果:
32.本发明以大肠杆菌和罗伊氏乳杆菌穿梭质粒pc2为基础,根据所要敲除的基因的数量及序列设计minicrispr序列,克隆至所述穿梭质粒pc2,构建基于罗伊氏乳杆菌的多基因编辑质粒pc2-mini(n)。基于罗伊氏乳杆菌的crispr系统进行基因编辑,不需要克隆外源性的核酸酶基因,即可实现罗伊氏乳杆菌的基因编辑,操作更为简单。
33.基于内源性crispr对罗伊氏乳杆菌进行基因编辑,通过点突变、缺失突变或插入突变改变其营养代谢途径或者插入新的代谢途径,从而达到对罗伊氏乳杆菌进行定向改造的目的,并且通过改造菌株的益生性能获得目的产物或者超表达目的产物,也可以阻断其不利代谢途径从而消除不利代谢产物的合成与分泌。
34.利用该技术进行基因编辑的效率远高传统的同源重组效率,且可以实现一次性的进行多个基因的编辑工作,不需要反复制作感受态细胞,大大缩短了对多个基因进行改造周期,节省时间成本。另外,本技术首次实现了在罗伊氏乳杆菌中进行基因的无痕整合,有利于将其开发为活体药物递送系统,发挥其作为肠道土著菌的优势。
35.利用crispr系统对细菌进行基因编辑,存在效率问题,即存在未发生基因编辑的菌株。通过该技术可以有效的实现对罗伊氏乳杆菌的编辑,编辑效率可达75%,针对未成功发生基因编辑的菌株,可以通过抗性培养条件下,传1-2代的方法获得成功发生基因编辑的菌株,解决了有些菌特别是乳酸菌转化效率普遍不高的问题。通过该方法,即使有些乳酸菌转化效率很低,但通过对获得的转化子进行传代,即可筛选得到发生基因编辑的菌株。
36.本发明完成编辑的菌株只需1-2次传代即可完成反筛,获得质粒丢失的目的菌株单克隆,从而进行第二次的基因编辑,有利于短时间内完成多个基因的连续编辑,获得在宿主肠道中益生性能更优良的目的菌株。
附图说明
37.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
38.图1为本发明质粒pc2的图谱;
39.图2为本发明mini crispr克隆到基因组上的测序结果;
40.图3为本发明质粒pc2-mini的图谱;
41.图4为本发明质粒pc2-s1s2-l1r1l2r2的图谱;
42.图5为本发明利用电转化方法,将编辑质粒pc2-mini转入罗伊氏乳杆菌,挑板子进行pcr验证,8个转化子均转入pc2-mini质粒;m为maker,1-8为8个转化子;
43.图6为本发明利用敲除质粒pc2-s-lr对罗伊氏乳杆菌进行单个基因敲除的方法获
取的缺失的杂合型敲除菌株,m为maker,1-8为敲除菌株,9为野生型菌株作为对照;
44.图7为将杂合型lacz缺失菌株进行划线挑单菌落获得的lacz纯合敲除株;m为maker,1-2为野生型与敲除型杂合株,3-4为纯合型lacz敲除株;
45.图8为本发明插入质粒pc2-s-g的图谱;
46.图9为本发明利用插入质粒pc2-s-g对罗伊氏乳杆菌lacz敲除株进行基因插入的方法获取的gfp基因插入的杂合型突变菌株;m为maker,1-8代表获取的8个不同的样品;其中5#和6#为发生插入的杂合突变型菌株;
47.图10为将杂合型gfp插入菌株进行划线挑单菌落获得的gfp纯合整合株,m为maker,1-23为挑取的不同单菌落样品,24为野生型菌株;其中,22#为纯合gfp插入菌株,23#为纯合lacz敲除菌株;
48.图11为本发明将已经成功转入基于罗伊氏乳杆菌基因编辑质粒pc2-s1-l1r1,却没有成功发生基因编辑的菌株进行传代,获取发生基因编辑的菌株;m为maker,1-8,10为成功转入pc2-s1-l1r1却没有成功发生基因编辑的不同菌株;其中,9为发生基因敲除的菌株;
49.图12为挑24个未成功发生敲除的单菌落用yzδlacz-f/yzδlacz-r验证,验证后代是否为成功发生敲除的菌株;m为maker,1-24为24个单菌落;其中19#菌株成功发生敲除,为杂合型敲除株;
50.图13为本发明对已完成编辑的罗伊氏乳杆菌基因工程菌进行反筛获取的插入gfp基因的纯基因型突变菌株且chl基因丢失的菌株;m为maker,1-8代表选取的反筛的8个样品,其中4#、8#为质粒丢失菌株。
具体实施方式
51.现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
52.应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
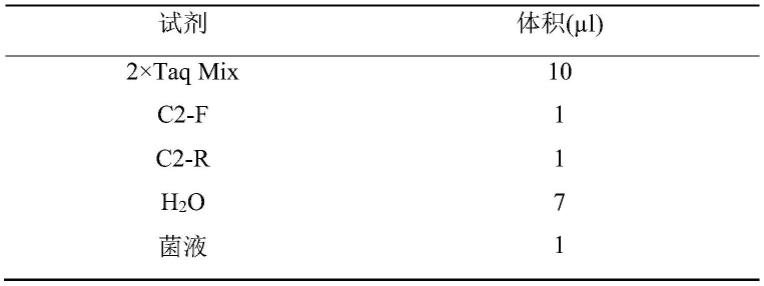
53.除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
54.在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本技术说明书和实施例仅是示例性的。
55.关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
56.实施例1罗伊氏乳杆菌基因编辑质粒的构建方法,具体包含以下步骤:
57.a、通过pcr反应得到含有gttttagatgtacttcaaatcaataatgtttagaac的两个重复序列以及两个重复序列中间加有两个三型酶切位点bsai的minicrispr片段;(多基因编辑质粒minicrispr的构建方法包括将n个重复序列以及每两个重复序列之间加入30bp根据所要靶标的基因的spacer,该序列由基因合成公司合成);
58.b、将minicrispr片段、pc2(实验室保藏,图谱如图1所示)分别进行酶切(37℃、6h),酶切位点为saci、xbai;
59.将上述酶切产物酶连转化至大肠杆菌,涂布含有10μg/ml氯霉素的lb固体培养基平板,37℃培养,挑取转化子并进行pcr验证;设计一对pc2的通用引物c2-f/c2-r,如下:
60.c2-f:5
’‑
ctcatcctcttcatcctcttcgtcttg-3’;
61.c2-r:5
’‑
gatctcaacaatgtgaagtcagctgcc-3’。
62.利用上述引物进行pcr扩增,反应体系如表1:
63.表1反应体系
[0064][0065]
pcr扩增反应条件为95℃预变性5min,95℃变性0.5min,59℃退火0.5min,72℃延伸0.5min(变性、退火、延伸过程循环34次),72℃反应5min即可。将pcr产物跑胶验证大小并送公司测序检测minicrispr片段是否克隆至质粒上,测序结果如图2所示(上、下两个图分别为克隆minicrispr到a、b两段序列前、后的测序结果);
[0066]
c、用含有10μg/ml氯霉素的lb液体培养基,在37℃、180rpm培养条件下活化上述正确转化子,抽提质粒得到基于罗伊氏乳杆菌的基因编辑质粒pc2-mini(图谱如图3所示)。
[0067]
实施例2制备感受态罗伊氏乳杆菌的方法,包括以下步骤:
[0068]
a、取冻存管中的罗伊氏乳杆菌接种到液体mrs培养基过夜培养,划线至mrs固体平板,于37℃培养箱中静置培养24h,长出单菌落后挑取至10ml mrs液体培养液中,于37℃静置培养培养12h;
[0069]
b、取1ml培养液转接至100ml新鲜mrs培养液中(添加有10ml的15%的甘氨酸),37℃、静置培养约6h至od
600
为1.2-1.3;
[0070]
c、取50ml菌液4℃、5500rpm离心5min,弃上清(重复至100ml菌体收完),再加入提前4℃预冷的20ml电转缓冲液i(蔗糖205.37g/l,三水磷酸钾1.86g/l,六水氯化镁0.20g/l,ph7.5)悬浮菌体,4℃、5500rpm离心5min,取上清(重复三次);
[0071]
d、取2ml提前4℃预冷的电转缓冲液ii(蔗糖171.15g/l,甘油10%)悬浮细胞,每80μl分装得到罗伊氏乳杆菌电转感受态,-80℃保存。
[0072]
实施例3基于罗伊氏乳杆菌的编辑质粒pc2-mini转化至感受态的方法,包括以下步骤:
[0073]
a、将500ng的pc2-mini加至一支80μl罗伊氏乳杆菌感受态细胞中,放冰上静置5min;
[0074]
b、将罗伊氏乳杆菌的感受态细胞和质粒混合物转移到提前预冷的电转杯中,放冰上静置5min;
[0075]
c、设置电转仪的电转参数为1500v、25uf、200ω,然后进行电转化;
[0076]
d、电转化完成后立即向电转杯中加入900μl预冷的复苏培养基,然后将电转杯中混合液取出到1.5ml离心管当中,37℃静置孵育6h;
[0077]
上述复苏培养基包含以下质量浓度的组分:蔗糖171.15g/l,葡萄糖20g/l,胰蛋白胨10g/l,酵母提取物4g/l,牛肉膏8g/l,乙酸钠3g/l,柠檬酸氢二铵2g/l,磷酸氢二钾2g/l,七水硫酸镁0.2g/l,一水硫酸锰0.05g/l,吐温80l ml/l;
[0078]
e、将孵育过后的电转化感受态细胞从培养箱中取出,4000rpm离心5min,收集菌体涂布含有5μg/ml氯霉素的mrs固体培养基平板,37℃培养;
[0079]
f、36h后平板上长出多个转化子,转化效率达到1
×
103~3
×
103cfu/ug;挑取8个转化子活化以后,用pc2上一对通用引物c2-f/c2-r:
[0080]
c2-f:5
’‑
ctcatcctcttcatcctcttcgtcttg-3’;
[0081]
c2-r:5
’‑
gatctcaacaatgtgaagtcagctgcc-3’;
[0082]
利用上述通用引物进行pcr扩增,检测是否含minicrispr的条带,pcr验证结果如图5所示:结果表明8个罗伊氏乳杆菌转化子均已转入编辑质粒pc2-mini,随机挑2#送测序,测序结果正确。
[0083]
实施例4利用基于罗伊氏乳杆菌单基因敲除质粒pc2-s-lr对其进行单个基因敲除的方法,包括以下步骤:
[0084]
a、以β-半乳糖苷酶lacz为待敲除基因,根据3’ngg设计gtcattgctaatttaacatctttatcagga为spacer1(lacz),将其分别通过bsai单酶切、酶连的方法克隆至pc2-mini,构建敲除干涉质粒pc2-s1;
[0085]
b、以罗伊氏乳杆菌基因组为模板,设计四对引物,即:
[0086]
lacz 1kl-f:gctctagacacttgaggttttacacatggctact;
[0087]
lacz 1kl-r:gattttgatatcgttcttgccgttagttaaagagccaacagaa ct;
[0088]
lacz 1kr-f:tgttggctctttaactaacggcaagaacgatatcaaaatcagc ag;
[0089]
lacz 1kr-r:aactgcagtccttgctgtgcgaccaatg。
[0090]
通过pcr的方法扩增lacz基因的上游和下游1000bp分别作为左臂、右臂,通过soe的方法将lacz-l与lacz-r连接起来,通过xbai/psti双酶切、酶连的方法克隆至pc2-s1,构建敲除质粒pc2-s1-l1r1;
[0091]
c、将敲除质粒pc2-s1-l1r1通过实施例3所述的转化方法转化至罗伊氏乳杆菌,用含有5μg/ml氯霉素的mrs平板进行筛选,挑取单菌落并进行pcr验证,设计基因组上lacz基因左臂右臂两端外侧的一对引物yzδlacz-f/yzδlacz-r,扩增基因组上包含lacz基因在内的一段序列,即:
[0092]
yzδlacz-f:ttggtaacagtcttaacattaacggatg;
[0093]
yzδlacz-r:aatggtggtaaatatcttcgtccag;
[0094]
利用验证引物进行pcr验证lacz是否发生缺失,分别得到两条大小不同的条带,判
断可知得到混合基因型菌株。如图6所示:图中1-9泳道为引物yzδlacz-f/yzδlacz-r的pcr验证结果,有敲除带(为杂合型),说明利用多基因敲除质粒pc2-s1-l1r1实现了对lacz基因的敲除,得到的菌株均为混合基因型菌株(9#为野生型菌株作为对照);
[0095]
d、将上述混合基因型菌株划线至不含抗生素的mrs平板,挑取单菌落,再次划线,挑取4个罗伊氏乳杆菌单菌落并活化,利用引物yzδlacz-f/yzδlacz-r,进行pcr验证,pcr验证结果如图7所示:图中第1泳道为maker,2-5泳道为引物yzδlacz-f/yzδlacz-r敲除菌株,说明获得的3#单菌落为lacz缺失的纯基因型敲除菌株。
[0096]
实施例5
[0097]
利用基于罗伊氏乳杆菌多基因编辑质粒pc2-ns-nlr对其进行多基因敲除的方法,包括以下步骤:
[0098]
a、以β-半乳糖苷酶lacz、lacl为待敲除基因,根据3’ngg设计gtcattgctaatttaacatctttatcagga为spacer1(lacz),atattcggccaattgttaccgataattatc为spacer2(lacl)将其分别通过bsai,bbsi单酶切、酶连的方法克隆至pc2-mini(n=2),构建多基因敲除干涉质粒pc2-s1s2;
[0099]
b、以罗伊氏乳杆菌基因组为模板,设计四对引物,即:
[0100]
lacz 1kl-f:gctctagacacttgaggttttacacatggctact;
[0101]
lacz 1kl-r:gattttgatatcgttcttgccgttagttaaagagccaacagaact;
[0102]
lacz 1kr-f:tgttggctctttaactaacggcaagaacgatatcaaaatcagcag;
[0103]
lacz 1kr-r:aactgcagtccttgctgtgcgaccaatg;
[0104]
lacl 1kl-f:gctctagaggagcctttgttgatattggtgt;
[0105]
lacl 1kl-r:ttgatcaataaagtctgataaatccctcctcgttactatctaca;
[0106]
lacl 1kr-f:aggagggatttatcagactttattgatcaagccatccttgt;
[0107]
lacl 1kr-r:aactgcagctccgctgtatatggtaaacaact。
[0108]
通过pcr的方法扩增lacz、lacl基因的上游和下游1000bp分别作为左臂、右臂,通过soe的方法将lacz-l与lacz-r、lacl-l与lacl-r连接起来,通过xbai/psti双酶切、酶连的方法克隆至pc2-s1s2,构建多基因敲除质粒pc2-s1s2-l1r1-l2r2(如图4所示);
[0109]
f、将敲除质粒pc2-s1s2-l1r1-l2r2通过实施例3所述的转化方法转化至罗伊氏乳杆菌,用含有5μg/ml氯霉素的mrs平板进行筛选,挑取单菌落并进行pcr验证,设计基因组上lacz、lacl基因左臂右臂两端外侧的一对引物yzδlacz-f/yzδlacz-r和yzδlacl-f/yzδlacl-r,扩增基因组上分别包含lacz、lacl基因在内的一段序列,即:
[0110]
yzδlacz-f:ttggtaacagtcttaacattaacggatg;
[0111]
yzδlacz-r:aatggtggtaaatatcttcgtccag;
[0112]
yzδlacl-f:tccattattacgccatgatgtaatgac;
[0113]
yzδlacl-r:gagttcttcgatgtgtgttgcat。
[0114]
利用验证引物进行pcr验证lacz、lacl是否发生缺失,分别得到两条大小不同的条带,判断可知得到混合基因型菌株,均有双敲除带,说明利用多基因敲除质粒pc2-s1s2-l1r1-l2r2实现了对lacz、lacl基因的敲除,得到的菌株均为混合基因型菌株;
[0115]
d、将上述混合基因型菌株划线至不含抗生素的mrs平板,挑取单菌落,再次划线,挑取4个罗伊氏乳杆菌单菌落并活化,利用引物yzδlacz-f/yzδlacz-r、yzδlacl-f/yzδ
lacl-r,进行pcr验证,得到lacz、lacl缺失的纯基因型敲除菌株。
[0116]
实施例6
[0117]
利用插入质粒pc2-s-g对罗伊氏乳杆菌进行基因插入的方法,包括以下步骤:
[0118]
a、以荧光蛋白基因gfp为待插入基因,根据3’ngg设计待插入位点lacz基因上一段gtcattgctaatttaacatctttatcagga序列为spacer,将其通过bsai单酶切、酶连的方法克隆至pc2-mini,构建插入质粒pc2-s;
[0119]
b、以罗伊氏乳杆菌基因组为模板,设计两对引物,通过pcr的方法扩增基因组上lacz基因位点的上下游1000bp分别作为左臂、右臂,通过soe的方法将左臂、gfp基因以及右臂连接起来,并通过xba1/psti双酶切、酶连的方法克隆至pc2-s,构建插入质粒pc2-s-g(图谱如图8所示);
[0120]
c、将插入质粒pc2-s-g通过实施例3所述的转化方法转化至罗伊氏乳杆菌lacz敲除株,用含有5ug/ml氯霉素的mrs固体培养基平板进行筛选,挑取单菌落,利用验证敲除lacz的引物yzδlacz-f/yzδlacz-r,即:
[0121]
yzδlacz-f:ttggtaacagtcttaacattaacggatg;
[0122]
yzδlacz-r:aatggtggtaaatatcttcgtccag,进行pcr扩增,结果如图9所示,验证了8个转化子,5#和6#的pcr结果为两条大小不同的条带,判断即为混合基因型菌株。
[0123]
d、将上述混合基因型菌株划线至不含抗生素的mrs平板,挑取单菌落,再次划线,挑取7个单菌落活化并利用引物yzδlacz-f/yzδlacz-r,进行pcr验证,结果图10所示,22#单菌落为lacz基因缺失、23#单菌落为gfp基因插入的纯基因型突变菌株、24#为野生型对照菌株。
[0124]
实施例7
[0125]
利用编辑质粒pc2-s-m对罗伊氏乳杆菌进行基因突变的方法,包括以下步骤:
[0126]
a、以罗伊氏乳杆菌lacl基因为待突变基因,根据3’ngg设计atattcggccaattgttaccgataattatc为spacer,将其通过bsai单酶切、酶连的方法克隆至pc2-mini,构建点突变干涉质粒pc2-s(m);
[0127]
b、以罗伊氏乳杆菌基因组为模板,设计两对引物,突变位点设计在引物上,通过pcr的方法扩增基因组上lacl基因突变位点的上下游1000bp分别作为左臂、右臂(突变位点包含在一个臂内,通过点突变一个碱基,在lacl基因中间产生了saci酶切位点),通过soe的方法将左臂、右臂连接起来,并通过酶切、酶连的方法克隆至pc2-s,构建突变质粒pc2-s-m;
[0128]
c、将突变质粒pc2-s-m通过实施例3所述的转化方法转化至罗伊氏乳杆菌,用含有5μg/ml氯霉素的mrs平板进行筛选,挑取单菌落;
[0129]
d、选取上述单菌落划线至不含抗生素的mrs平板,挑取单菌落,再次划线,挑取10个单菌落活化并对lacl基因进行pcr扩增,然后进行saci酶切,跑胶,成功的发生点突变的菌株所扩增的pcr产物会被saci切割,产生小的条带。
[0130]
实施例8
[0131]
从含有编辑质粒却未成功发生基因编辑的罗伊氏杆菌出发,获得发生基因编辑菌株的方法,具体包括以下步骤:
[0132]
将已经成功转入基于罗伊氏乳杆菌基因编辑质粒pc2-s1-l1r1,却没有成功发生基因编辑的菌株(图11中的8#)在含有抗生素的液体培养基中进行传代培养,再将长好的菌
液划线至抗性平板,然后挑24个单菌落用yzδlacz-f/yzδlacz-r验证,发现后代有成功发生敲除的菌株(图12中的19#)。
[0133]
实施例9
[0134]
对已完成编辑的罗伊氏乳杆菌进行反筛的方法,具体包括以下步骤:
[0135]
将已完成lacz基因敲除的混合基因型罗伊氏乳杆菌(图9中的4#)在不含抗生素的mrs平板上划线进行传代培养,重复该过程1次,挑取平板上的单菌落8个,利用引物:
[0136]
chl-f:aatgtggtctttattcttcaactaaagc,
[0137]
chl-r:ttataaaagccagtcattaggcct,扩增质粒上的chl基因,pcr扩增结果如图13所示:4#、8#单菌落为lacz基因敲除的纯基因型突变菌株,且chl基因丢失,即发生敲除质粒丢失。
[0138]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1