一种高通量单细胞转录组测序方法及其应用
1.本发明属于生物技术领域,涉及单细胞测序领域,具体而言涉及一种多类型细胞的单细胞转录组测序方法及其应用。
背景技术:
2.目前使用最广泛的高通量单细胞转录组测序技术是10x genomics公司开发的基于液滴微流控平台的单细胞测序技术,该技术可实现数千个细胞的标记、测序和分析,获得单细胞水平的基因表达谱,实现细胞亚群的划分和细胞亚群间差异表达基因的检测,与之类似的技术还有indrop技术和drop-seq技术。
3.10x genomics转录组测序技术的基本操作和原理为(参见图1):将样本制备成单细胞悬浮液(其中的细胞活性需高于90%,细胞浓度一般约为700~1200个细胞/μl),之后通过微流控平台将带有条形码(barcode)和引物的单个编码微球(编码微球由凝胶珠和凝胶珠上连接的一段引物构成)与单个单细胞包裹在油滴中;在每个单个液滴中,凝胶珠溶解,细胞裂解释放mrna,通过反转录产生用于测序的带条形码和umi信息的cdna;破碎液滴油层后收集cdna进行扩增,制备cdna文库,然后使用illumina测序平台对文库进行测序检测,从而获得大量单细胞的基因表达数据。
4.上述测序技术中使用的凝胶珠上连接的引物序列包含四个部分:illumina truseq read 1测序引物、16nt的条形码、12nt的umi和30nt的poly(dt)反转录引物,其中truseq read 1测序引物为一段已知的短肽核苷酸序列,用于后续的上机测序;条形码则与微珠一一对应,共有400万种条形码;umi是由随机碱基组成的一段序列,凝胶珠上每个dna分子都有自己的umi序列,其作用在于混合测序时区分不同的转录本(即区分哪些测序得到的序列是来自于同一个原始cdna分子);poly(dt)反转录引物则是含有30个t碱基的同聚dna片段,用于捕获有polya尾的转录本。
5.以10x genomics为代表的高通量单细胞转录组测序技术中使用的反转录引物均为poly(dt)反转录引物,因此只能获得部分3’端转录本信息,并且无法检测到不含有polya的rna(包括损伤的mrna片段和不含polya的mirna、lncrna等),这导致该技术在实际应用中灵敏度很低,通常只有少于10%的mrna可被检测到。同时,该技术对rna质量的要求较高,如果想要获得较好的测序结果,测序样本的细胞活率大于80%,采用冻存或固定样本时测序的效果往往很差。此外,由于细菌的rna不带有polya,现有的高通量单细胞转录组测序技术因而无法用于细菌的转录组测序。同时,该技术中为了避免污染,在进行细胞分隔时需要尽可能使得每个液滴中仅包括一个细胞,为此在制备过程中液滴量远远高于细胞的量并且通常每次仅对一种类型的单细胞进行测序,这导致最终只有约1/10的液滴有细胞,大量空液滴中的微球和试剂被浪费。
6.因此,本领域中存在的对于捕获效率更高、成本及污染率更低、细胞样本通量更高的单细胞转录组测序方法的需要。
技术实现要素:
7.为了解决上述问题,在第一个方面,本发明的目的在于提供一种单细胞转录组测序方法,所述方法包括将待测的细胞样本制备成单细胞悬液后用固定液固定;和使用反转录引物在固定后的单细胞的rna上进行原位反转录反应合成cdna第一条链;其中所述反转录引物中包括标签序列。
8.在具体的实施方案中,本发明的方法中所述反转录引物可以包含标签序列和rna结合序列,所述rna结合序列可以是随机rna结合序列、针对目标rna序列设计的rna结合序列或其组合。
9.在具体的实施方案中,本发明的方法还可以包括在反转录获得的单细胞的cdna第一条链的尾端加上相应的捕获接头,所述捕获接头与编码微球上的捕获接头互补链的序列互补;所述捕获接头是已知序列的任意片段,优选地所述捕获接头是poly(da)片段,poly(dt)片段,poly(dg)片段或poly(dc)片段。
10.在具体的实施方案中,本发明的方法还可以包括将单细胞和单个所述编码微球包含在单个腔室中以形成细胞分隔;和在形成所述细胞分隔后在所述单个腔室中合成cdna第二条链;其中形成所述细胞分隔后至少部分所述单个腔室中包含两个或两个以上单细胞。
11.在具体的实施方案中,本发明的方法中所述两个或两个以上单细胞可以为相同类型或不同类型的细胞。
12.在具体的实施方案中,本发明的方法还可以包括在合成所述cdna第二条链后对双链cdna进行pcr扩增及建库和测序。
13.在具体的实施方案中,本发明的方法使用的编码微球上的单链dna中可以包含上游扩增引物互补片段、条形码、umi和捕获接头互补链。在优选的实施方案中,所述条形码可以是一个或多个条形码;在更优选的实施方案中,所述条形码可以是三个条形码。
14.在具体的实施方案中,本发明的方法中可以使用微流控芯片或微孔板完成所述细胞分隔。
15.在具体的实施方案中,本发明的方法中所用的固定液可以是单纯固定液或混合固定液;优选地所述单纯固定液可以包括但不限于多聚甲醛、甲醛、福尔马林、甲醇、丙酮、乙醇、醋酸、苦味酸、铬酸、重铬酸钾和升汞;优选地所述混合固定液可以包括但不限于醋酸-酒精混合液、福尔马林-醋酸-酒精液和包因氏固定液。
16.在具体的实施方案中,本发明的方法可以包括:将待测的细胞样本制备成单细胞悬液后用固定液固定;使用反转录引物在固定后的单细胞的rna上进行原位反转录反应合成cdna第一条链,所述反转录引物从5’端至3’端依次包括下游扩增引物互补片段,标签序列和rna结合序列;在反转录获得的cdna第一条链的尾端加上捕获接头,所述捕获接头与编码微球上的捕获接头互补链的序列互补;将所述单细胞和单个所述编码微球包含在单个腔室中以形成细胞分隔,其中形成所述细胞分隔后至少部分所述单个腔室中包含两个或两个以上单细胞;在所述单个腔室中合成cdna第二条链以形成双链cdna;和对所获得的双链cdna进行pcr扩增及建库和测序。
17.在第二个方面,本发明的目的在于提供第一个方面中的方法在单细胞、单细胞核、单个微生物的全转录组测序中的应用,优选地所述应用可以是微生物学、基础医学、临床医学、农学、细胞生物学、免疫学、发育生物学、病理学、神经生物学及发育、遗传学、干细胞、肿
瘤、生殖健康、宏基因组学及微生态、新药研发领域中的应用。
18.与现有技术相比,本发明的测序方法具有以下优点:
19.(1)本发明的方法可测量的细胞样本的类型更广,可应用于真核细胞的单细胞和单细胞核、原核生物(细菌、放线菌、立克次氏体、衣原体、支原体、蓝细菌和古细菌等)、单细胞藻类、病毒等;并且不仅可适用于新鲜样本,也可应用于细胞活性较低的固定样本、冰冻保存样本、石蜡包埋样本(ffpe)等;
20.(2)本发明的方法可以检测编码rna和多种形式的非编码rna并获得更加完整的转录组图谱,进行转录组水平定量、基因差异表达、交替剪接、基因融合、rna互作等分析;
21.(3)对比目前的微流控单细胞rna测序技术,在同等测序深度下本发明的方法的rna检测灵敏度更高;
22.(4)本发明的方法单次可检测的细胞样品通量更高,并且可以同时对多种类型细胞进行单细胞转录组的测序,在保证测序精确度和灵敏度的同时有效减低检测时间和经济成本。
附图说明
23.图1是现有的10x genomics单细胞转录组测序方法的原理示意图。
24.图2是本发明的单细胞转录组测序方法的原理示意图。
25.图3是本发明中使用的编码微球的结构示意图。
26.图4是采用本发明方法处理大肠杆菌样本并进行测序的结果。a.显微镜下溶菌酶降解细胞壁后的大肠杆菌;b.显微镜下反转录反应后的大肠杆菌;c.qpcr实验的扩增曲线;d.核酸凝胶电泳图;e.大肠杆菌中的基因检出数。
27.图5是采用本发明方法对大肠杆菌和枯草芽孢杆菌的混合样本进行测序的结果;单细胞中比对到不同菌种基因组上的umi数目散点图,其中umi数即为测序计数出来的cdna分子数目,图中每个点代表一个细胞,浅色的点代表的细胞中几乎只含有枯草芽孢杆菌的cdna,深色的点代表的细胞中几乎只含有大肠杆菌的cdna,黑色的点代表的细胞为有污染的细胞。
28.图6是采用本发明方法对人鼠细胞系混合样本进行测序的结果;其中a.采用本发明的方法对人鼠细胞系混合样本测序得到的小鼠单细胞基因检出数;b.采用现有10x genomics等七种测序方法对人鼠细胞系混合样本测序得到的小鼠单细胞基因检出数;c.测序结果中小鼠单细胞基因的测序读长(reads)在参考基因组不同区域的分布情况;d.测序结果中小鼠单细胞基因的测序读长在参考基因5'-3'上的分布均匀性统计图。
29.图7是采用本发明方法对人鼠细胞系混合样本进行测序的结果。其中a是显微镜下细胞形态,b是文库大小分布范围,c是测序细胞read和umi数量分布,d是测序结果中人鼠细胞umi污染分布统计。
30.图8是采用本发明方法对小鼠肝脏组织石蜡包埋组织(ffpe)样本进行测序的结果;其中a.显微镜下解离后的小鼠肝脏组织细胞核;b.显微镜下反转录反应后的小鼠肝脏组织细胞核;c.qpcr实验的扩增曲线;d.核酸凝胶电泳图;e.小鼠肝脏组织细胞核中的基因检出数。
31.图9是采用本发明方法对烟草细胞核样本进行测序准备的结果。a.显微镜下解离
后的植物细胞核;b.显微镜下反转录反应后的植物细胞核;c.qpcr实验的扩增曲线;d.核酸凝胶电泳图。
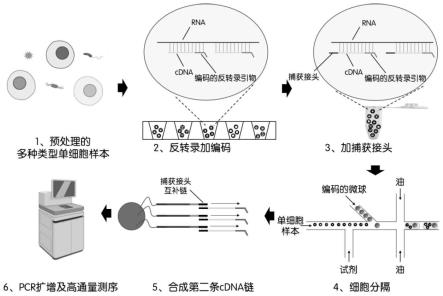
32.图10是采用本发明方法对衣藻和蓝藻样本进行测序准备的结果。a.显微镜下降解细胞壁后的衣藻和蓝藻;b.显微镜下反转录反应后的衣藻和蓝藻;c.qpcr实验的扩增曲线;d.核酸凝胶电泳图。
33.图11是采用本发明方法对不同类型固定液固定的细胞样本进行测序准备的结果;a.显微镜下反转录反应后的3t3细胞;b.qpcr实验的扩增曲线;c.核酸凝胶电泳图。
34.图12是采用本发明方法对cdna链尾端加不同捕获接头片段的细胞样本进行测序准备的结果;其中a.qpcr实验的ct值;b.核酸凝胶电泳图。
35.图13是采用本发明方法对小鼠脑组织样本进行测序的结果。其中a是使用方案一和方案二对样本进行测序所获得的基因数,read数和线粒体基因分布。b是两种方法脑组织的分群和高表达基因热图。c是两种方法脑组织分群的相关性分析。
36.发明详述
37.本发明的实施方案提供一种单细胞转录组全rna测序方法(如图2所示),该方法的具体步骤包括:对细胞样本进行预处理;使用反转录引物在固定后的单细胞上结合目标rna发生原位反转录反应,所述反转录引物中包含标签序列;之后在反转录合成的第一条cdna链尾端加上捕获接头;随后制成包含单个编码微球、单细胞和反应试剂的单个腔室;在单个腔室中编码微球通过捕获接头互补链与第一条cdna结合并进行延伸反应,合成带有条形码标记的cdna第二条链;最后对获得的双链cdna进行pcr扩增和高通量测序。本技术的测序方法允许在细胞分隔步骤中形成包含两个或两个以上单细胞的单个腔室,从而建立高通量高灵敏度的单细胞转录组测序平台。
38.定义
39.本文中的单细胞包括但不限于真核细胞的单细胞/单细胞核、原核生物(细菌、放线菌、立克次氏体、衣原体、支原体、蓝细菌和古细菌等)、单细胞藻类、病毒等。
40.本文中的细胞样本包括但不限于细胞的固定样本、冰冻保存样本、石蜡包埋样本(ffpe)等。
41.本文中的rna包括能检测的编码rna和多种形式的非编码rna,例如mirna、lncrna、sirna、circrna等。
42.本文中的不同类型的细胞可以是不同物种的细胞,也可以是相同物种的细胞(例如不同培养批次的细胞、不同来源的细胞)。
43.本发明的反转录引物至少包含标签序列以及rna结合序列,所述rna结合序列可与目标rna序列结合,rna结合序列可以是随机rna结合序列,或者是针对特定基因设计的rna结合序列,也可以它们的组合,并且在实际操作中,反转录引物中可以适当加入poly(dt)引物。所述标签序列可以在读取测序数据时区分不同细胞,其可以是条形码片段或其他可以区分不同细胞的碱基序列。此外,反转录引物中还可以包含下游扩增引物互补片段,以便需要时在后续pcr扩增反应中与下游扩增引物互补结合。在本发明的实施例中反转录引物从5’至3’端依次包括下游扩增引物互补片段、标签序列和6个随机碱基序列(即5
’‑
下游扩增引物互补片段扩增-标签序列-nnnnnn-3’,n=dg,da,dt或dc)组成,其中6个随机碱基序列可与目标rna序列结合,下游扩增引物互补片段则用于在pcr扩增反应中与下游扩增引物结
合。为了区别,本技术中的“反转录随机引物”也是可以用于反转录步骤的引物,其与反转录引物的区别在于不包含标签序列,在本发明的实施例中反转录随机引物从5’至3’端依次包括下游扩增引物互补片段和6个随机碱基序列(即5
’‑
下游扩增引物互补片段-nnnnnn-3’,n=dg,da,dt或dc)组成,其中6个随机碱基序列可与目标rna序列结合,下游扩增引物互补片段则用于在pcr扩增反应中与下游扩增引物结合。
44.本文中的条形码(barcode)片段是指一串用于区分不同细胞的碱基序列,作为细胞标签,要求这段碱基序列是稳定的、可合成的、且变异性高的。一般可自行设计或可在条形码库中选择合适的条形码片段。目前市场上主流的两款单细胞测序技术10x genomics和新格元的条形码库可以分别在其开源的定量软件cellranger和celescope中查到。
45.下文中将对本发明测序方法中的主要步骤进行概括性描述。
46.编码微球的准备
47.本发明中的编码微球上的单链dna由上游扩增引物互补片段、条形码、umi(unique multiplex index)和捕获接头互补链组成(参见图3)。上游扩增引物互补片段在pcr扩增反应中与上游扩增引物结合;条形码用于对同一细胞分隔中的cdna进行标记;umi为一段随机序列,用于标记每一个原始cdna;捕获接头互补链在cdna第二条链合成反应中与连接在第一条cdna链上的捕获接头结合。本发明的实施方案中的编码微球可以采用10x genomics、1cellbio、新格元、bd rhapsody等的单细胞测序试剂盒中的微球。
48.样本预处理
49.制备单细胞悬液:可对应不同类型的细胞样本选择相应的消化酶,以将细胞样本制备成单细胞悬液。比如对于培养细胞可用胰酶/edta消化成单细胞;对于新鲜组织可用相应的消化酶(如肌肉组织选择胶原酶ⅰ和分散酶、肝脏组织选择胶原酶ⅳ)消化后经过滤、洗涤制备成单细胞;对于冰冻保存样本需要先将样本在25~60℃水浴锅中快速融化;对于石蜡包埋样本(ffpe)需要先用二甲苯或其他环保型脱蜡剂脱蜡,再进行解交联;对于单细胞核转录组测序,需要先对单细胞样本用强非离子表面活性剂(np-40等)处理裂解细胞膜。
50.固定单细胞:当待测样品为单一细胞类型时,将待测细胞样本的单细胞悬液用固定液固定。当待测样品包含不同类型细胞时,可以将各个类型待测细胞样本的单细胞悬液分别用固定液固定后混合,并再进行后续反转录;或者可以将待测细胞样本混合后制成单细胞悬液并用固定液固定,并再进行后续反转录;或者还可以将各个类型待测细胞样本的单细胞悬液分别用固定液固定,并在进行后续反转录之后细胞分隔之前将不同类型细胞的样本混合。一般使用固定液处理样本,将细胞/细胞核内部大分子(rna、蛋白质等)结构固定,使其在后续实验过程中保持完整的单细胞/细胞核形态、结构及组成,并且rna能稳定地固定在细胞/细胞核内。操作时可根据不同样本类型特点,选择合适的固定液,固定液包括但不限于多聚甲醛、甲醛、福尔马林、甲醇、丙酮、乙醇、醋酸、苦味酸、铬酸、重铬酸钾、升汞等单纯固定液,以及醋酸-酒精混合液、福尔马林-醋酸-酒精液、包因氏固定液等混合固定液。以及不同的固定时间,例如15min~30min或者过夜。
51.反转录
52.在固定后的细胞的rna上通过多位点结合用于反转录的引物发生原位反转录反应,其中所使用的引物可以根据不同实际要求选择。当所加入的引物是不包括标签序列的反转录随机引物时,为了避免污染,细胞分隔后中单个腔室中仅包含一个单细胞。当所加入
的引物是包含标签序列的反转录引物时,细胞分隔后可允许至少部分单个腔室中包含两个或两个以上细胞,以在96孔板中进行的反转录为例,不同的孔中进行反转录,每个孔中加入携带特定标签序列的反转录引物,后续在细胞分隔步骤中携带不同标签序列的细胞可以被分在同一个细胞分隔中。
53.反转录体系中还可加入10%tritonx-10对细菌的细胞膜起通透作用,使反应试剂可以更容易地进入细菌内部。
54.加捕获接头
55.反转录后的第一条cdna链3’尾端需要加一个捕获接头使其能与编码微球单链cdna上的捕获接头互补链结合以进一步合成cdna第二条链。本发明的实施方案中可以通过末端转移法在第一条cdna链尾端加上poly(da)、poly(dt)、poly(dg)或poly(dc)作为捕获接头;也可以通过dna连接法在第一条cdna链尾端加上一段特定的捕获接头;也可以采用模板置换法,在反转录步骤使用反转录酶在反转录后的第一条cdna链尾端加上三个dc作为捕获接头。而相对地,编码微球单链cdna上的捕获接头互补链可作相应调整。
56.细胞分隔
57.细胞分隔是指形成包括单个编码微球、一个或多个单细胞、反应试剂的单个腔室。为了完成后续合成cdna第二条链的延伸反应,所述单个腔室中的延伸反应试剂通常包括dna聚合酶、dntps和反应缓冲液。可以根据不同细胞的大小和类型,设计不同的微流控芯片产生微液滴进行细胞分隔或使用微孔板(microwell)技术进行细胞分隔。一般根据测序所需检测的细胞数量收集相应数量的单液滴或者制备相应数量的微孔。最终单个腔室内的单细胞个数与细胞分隔过程中体系中的单细胞浓度有关,细胞浓度逐渐增加时,细胞分隔后空液滴比例逐渐降低,当细胞浓度达到一定数值后,细胞分隔后部分单个腔室中将包含两个甚至两个以上的单细胞。具体来说,与细胞分隔后单个腔室基本上只包含一个单细胞的细胞分隔体系相比,本发明的方法中细胞分隔体系中细胞浓度一般可提高至10-20倍,此时,细胞分隔后空液滴比例将大大减少,并且部分单个腔室中将包含两个甚至两个以上的单细胞。
58.合成cdna第二条链
59.编码微球连接的单链cdna上的捕获接头互补链与加在第一条cdna链上的捕获接头结合,随后在dna聚合酶作用下延伸合成cdna的第二条链。所获得的cdna的第二条链含有上游扩增引物互补片段、条形码、umi、捕获接头互补链以及cdna序列。
60.构建文库及高通量测序
61.首先用磁珠法纯化出上一步延伸反应后的原始双链cdna,加入上游引物和下游引物以及pcr扩增反应的试剂,对原始双链cdna进行pcr扩增。用磁珠法纯化pcr扩增产物,在pcr扩增产物两端连接上测序接头(adapter),例如可采用ta克隆连接接头建库法或pcr法。构建好的文库可用illumina测序平台或华大智造测序平台进行高通量测序。pcr扩增反应的上游引物和下游引物分别根据编码微球单链cdna上的上游扩增引物互补片段和反转录随机引物中的下游扩增引物互补片段设计并合成。
具体实施方式
62.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例
中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。
63.基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
64.下面结合附图并通过具体实施方式来进一步说明本发明的技术方案。
65.实施例1:大肠杆菌样本测序(细胞分隔后单个腔室中仅包含一个单细胞)
66.样本预处理
67.分别取约100万个不同菌株(1、2、3、4)大肠杆菌验证本发明专利可用于细菌样本的单细胞转录组测序,加入含0.05%tween-20(购于上海生工生物有限公司)的pbs缓冲液(pbst)洗三次,通过震荡和过滤去掉团聚的细菌,使样本中的细菌形成单细菌悬液,加入1%多聚甲醛(购于北京索莱宝科技有限公司),置于4℃固定过夜。固定结束的细菌样本用pbst缓冲液洗三次,加入溶菌酶(购于美国thermo fisher scientific公司)裂解细胞壁,37℃处理15分钟,加入pbst缓冲液洗三次。
68.反转录
69.预处理后的样本中加入含反转录酶(购于美国thermo fisher scientific公司)、反转录反应缓冲液、dntps(购于北京索莱宝科技有限公司)、反转录随机引物(由上海生工生物有限公司合成)、10%tritonx-10(购于上海生工生物有限公司)的反转录-通透反应试剂进行反转录反应,每50ul的反转录反应体系用于每50万单细菌。反应结束后加入pbst缓冲液洗三次。
70.加捕获接头
71.反转录后的样本中加入末端转移酶(购于美国thermo fisher scientific公司)、dctp(购于上海生工生物有限公司)和反应缓冲液,37℃孵育30分钟,使第一条cdna链的3’尾端加上poly(dc)捕获接头片段。反应结束后加入pbst缓冲液洗三次。显微镜下进行镜检,结果显示在裂解完细胞壁和反转录反应后,大肠杆菌样本仍可以保持分散、完整的单细菌形态(参见图4a-b)。
72.细胞分隔
73.对每个样本进行计数,调整其细菌浓度为200~400个/ul。将细菌样本、延伸反应试剂(包括dna聚合酶(购于美国thermo fisher scientific公司)、dntps和反应缓冲液)、编码微球(购于美国1cellbio公司)、油相(含0.2%表面活性剂的电子氟化液7500,购于美国3m公司)分别加入注射器中,通过软管分别与微流控芯片的对应入液口连接,设置合适的流速,形成包含单个细菌、单个编码微球、延伸反应试剂的油包水单液滴,收集约200ul的单液滴,形成单个腔室包含一个单细胞的细胞分隔。
74.合成cdna的第二条链
75.将收集的单液滴分装到不同管中后进行延伸反应,以在单液滴中合成带条形码标记的cdna第二条链。延伸反应结束后在各个管中加入20%pfo(含20%1h,1h,2h,2h-perfluorooctanol的电子氟化液7500,购于美国sigma-aldrich公司)以将单液滴破碎,通过磁珠法(磁珠购于美国beckman coulter公司)纯化提取管中的cdna。
76.构建文库及高通量测序
77.取部分双链cdna(200~400个细胞)作为模板进行qpcr实验检测捕获到的总cdna
含量,结果显示得到ct值约为17(参见图4c)。根据得到ct值计算合适的扩增循环数(一般取ct值加3个循环),将剩余其他管中的cdna样本进一步通过pcr扩增反应将编码的cdna扩增至合适的总含量(约100ng)。取3ul扩增产物进行核酸电泳,结果显示得到弥散状条带,条带大小分布主要为200~500bp(参见图4d),说明本发明方法从大肠杆菌样本中捕获的cdna捕获率高、覆盖范围广,可用于后续高通量测序。
78.取100ng扩增后的cdna采用ta克隆连接接头建库法进行末端修复和加a尾,连接上接头(建库试剂盒购于美国illumina公司)。构建好的文库用illumina测序平台进行高通量测序。测序结果显示,在单个大肠杆菌中可以测到基因数目分布在1000~3000(参见图4e),基本包含了大肠杆菌中表达的所有基因,说明本发明方法可应用于细菌的单细胞转录组测序,且灵敏度高。
79.实施例2:大肠杆菌和枯草芽孢杆菌混合样本测序(细胞分隔后单个腔室中仅包含一个单细胞)
80.取约100万个大肠杆菌和枯草芽孢杆菌混合样本验证应用本发明专利进行单细胞转录组测序的准确性。用上文实施例1中描述的方法进行样本预处理、反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库及高通量测序。测序结果显示,经过基因组比对后大肠杆菌和枯草芽孢杆菌混合样本中总共检测到251个细菌,其中有4个细菌中既含有大肠杆菌的cdna又含有枯草芽孢杆菌的cdna,这通常是由于两个细菌进入同一个液滴所造成的,其余的细菌中几乎只含有大肠杆菌cdna或含有枯草芽孢杆菌cdna,总体污染率大概小于2%(参见图5)。说明应用本发明专利进行单细胞转录组测序准确性高,可以很好地将样本中混合的大肠杆菌和枯草芽孢杆菌分开,物种间交叉污染少。
81.实施例3:人鼠细胞系混合样本测序
82.仅对小鼠3t3细胞进行测序(细胞分隔后单个腔室中仅包含一个单细胞):取约100万个-80℃冻存的人鼠细胞系混合样本(50%人hek293细胞和50%小鼠3t3细胞),取出冻存的细胞样本于37℃水浴锅中快速解冻,加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜。固定结束的细胞冻存样本加入pbst缓冲液洗三次。随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库及高通量测序。进行基因组比对后分离出小鼠3t3细胞的数据,结果显示本发明方法在冻存细胞样本中可以检测小鼠单细胞基因数目分布在4000~10000(参见图6a);文献systematic comparison of single-cell and single-nucleus rna-sequencing methods(单细胞和单细胞核rna测序方法的系统比较,jiarui ding等人,nat biotechnol.2020june;38(6):737
–
746.)中报道用七种现有技术单细胞转录组测序方法(七种测序方法包括2种基于微孔板的低通量方法(smart-seq2和cel-seq2)和5种高通量方法(10x chromium,drop-seq,seq-well,indrops和sci-rna-seq))对人鼠细胞系混合样本(50%人hek293细胞和50%小鼠3t3细胞)进行测序,将采用本发明方法检测的小鼠单细胞测序结果与上述七种方法检测的小鼠单细胞的测序数据相比,在相同测序深度下,本发明测序方法的灵敏度达到了smart-seq2等低通量单细胞测序方法,远高于10x genomics(10x chromium)等高通量单细胞测序方法(参见图6b)。测序比对结果显示小鼠单细胞的测序读长分布在参考基因组不同区域,包括编码rna、基金间区域、内含子间、非翻译区(参见图6c),说明采用本发明专利可以对全转录组进行测序;测序比对结果显示小鼠单细胞的测序读长在参考基因的5'-3'上分布均匀(参见图
6d)。
83.同时对人hek293细胞和小鼠3t3细胞进行测序(细胞分隔后至少部分单个腔室中包含两个或两个以上的单细胞):取约500万个-80℃冻存的人鼠细胞系混合样本(50%人hek293细胞和50%小鼠3t3细胞),取出冻存的细胞样本于37℃水浴锅中快速解冻,加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜。固定结束的细胞冻存样本加入pbst缓冲液洗三次(参见图7a)。随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库及高通量测序。其中反转录过程在96孔板中进行,每孔反应体系缩小为10ul,每孔使用的引物为携带不同的标签序列的反转录引物,从而使同一孔中细胞的cdna携带相同的标签序列,不同孔间细胞的cdna携带不同的标签序列。细胞分隔时使用10倍浓度的细胞样品(2000~4000个/ul),其他试剂和微球浓度不变,形成单个腔室包含多个单细胞的细胞分隔。文库大小控制在200-500bp(参见图7b)。进行基因组比对后分离出小鼠3t3和人293细胞的数据,结果显示本发明方法在人鼠混合实验中通过预标记方案可以检测到更多细胞,污染率控制在较低水平(1.2%),检测到转录本数量也可以达到6000左右(参见图7c-d)。
84.实施例4:小鼠肝脏组织石蜡包埋组织(ffpe)样本测序准备(细胞分隔后单个腔室中仅包含一个单细胞)
85.取约20mg小鼠肝脏组织的石蜡包埋样本,首先对石蜡包埋样本进行脱蜡和复水处理,加入细胞裂解液将组织裂解成单细胞核,加入pbst洗三次。随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库及高通量测序。结果显示,从ffpe样本中成功分离的单个分散细胞核在反转录反应后仍可以保持分散、完整的单细胞核形态(参见图8a,b)。获得纯化的双链cdna产物后,取部分cdna(约100个细胞核)作为模板进行qpcr实验检测捕获到的总cdna含量,得到ct值约为10(图8c),这表明本发明方法对ffpe样本单细胞核中cdna捕获率较高。根据ct值对cdna产物进行pcr扩增(15个循环),取3ul扩增产物进行核酸电泳,得到弥散状条带,条带大小分布主要为200~500bp(图8d),表明本发明方法对ffpe样本单细胞核中捕获到cdna捕获率高、覆盖范围广,可用于后续测序。测序结果显示,本发明方法在ffpe样本单细胞核中可以检测到2000~3000个基因(参见图8e),说明本发明方法可应用于ffpe样本的单细胞核转录组测序。
86.实施例5:烟草细胞核样本测序准备(细胞分隔后单个腔室中仅包含一个单细胞)
87.用单细胞制备仪将新鲜烟草样本解离成单细胞核(参见图9a),取约100万烟草细胞核样本,加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜。固定结束后将烟草细胞核样本用pbst缓冲液洗三次。随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库。结果显示,反转录反应后,烟草细胞核样本仍可以保持分散、完整的单细胞核形态(参见图9b)。qpcr实验结果得到烟草细胞核的ct值约为16(参见图9c),这表明本发明方法可以捕获较多的烟草细胞核样本中cdna。核酸凝胶电泳结果得到弥散状条带,条带大小分布主要为150~300bp(图9d),表明本发明方法从烟草细胞核样本单细胞中捕获到的cdna捕获率高、覆盖范围广,可用于后续测序。
88.实施例6:衣藻和蓝藻混合样本测序准备(细胞分隔后单个腔室中仅包含一个单细胞)
89.分别取约100万个衣藻和蓝藻样本验证本发明专利可用于衣藻和蓝藻样本的单细
胞转录组测序。衣藻和蓝藻样本中加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜。固定结束后将衣藻和蓝藻样本用pbst缓冲液洗三次。对于衣藻样本,加入含4%纤维素酶、0.5%果胶酶、0.3mol/l甘露醇,ph=6.5的细胞壁酶解液,37℃处理15分钟,加入pbst缓冲液洗三次。对于蓝藻样本,加入溶菌酶裂解细胞壁,37℃处理15分钟,加入pbst缓冲液洗三次。裂解完细胞壁的衣藻和蓝藻样本随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库。结果显示,在裂解完细胞壁和反转录反应后,衣藻和蓝藻样本仍可以保持分散、完整的单细胞形态(参见图10a、b)。qpcr实验结果得到衣藻样本的ct值约为17,蓝藻样本的ct值约为17(图10c),这表明本发明方法对衣藻和蓝藻样本中cdna捕获率均较高。核酸凝胶电泳结果得到弥散状条带,条带大小分布主要为200~500bp(图10d),表明本发明方法从衣藻和蓝藻样本单细胞中捕获到的cdna捕获率高、覆盖范围广,可用于后续测序。
90.实施例7:对小鼠3t3细胞系样本进行测序准备(采用不同固定液和不同捕获接头,细胞分隔后单个腔室中仅包含一个单细胞)
91.分别取4组培养的小鼠3t3细胞系验证应用不同固定液处理样本的效果。在细胞培养皿中加入胰酶在37℃消化3分钟,将其消化成单细胞,加入pbs缓冲液洗三次,分别加入1%多聚甲醛、70%乙醇、丙酮、1%醋酸(样本编号为1、2、3、4),置于4℃固定过夜,固定结束后加入pbst洗三次。随后参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库。结果显示不同类型固定液固定的细胞样本在反转录反应后均能保持完整的单细胞形态(参见图11a)。本发明方法对不同类型固定液固定的细胞样本的cdna捕获率均较高(参见图11b),得到弥散状条带,条带大小分布主要为200~500bp(参见图11c),说明本发明方法从不同类型固定液固定的细胞样本中捕获的cdna捕获率高、覆盖范围广,均可用于后续高通量测序。
92.另外分别取7组培养的小鼠3t3细胞系验证在cdna第一条链尾端加不同捕获接头的效果。在细胞培养皿中加入胰酶在37℃消化3分钟,将其消化成单细胞,加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜,固定结束后加入pbst洗三次。取6组固定后的细胞样本根据上文实施例2描述的方法进行反转录反应。取4组反转录结束后的细胞样本中分别加入datps,dttps,dgtps和dctps,以及末端转移酶和反应缓冲液,在37℃孵育30分钟,分别在cdna尾端加上poly(da)、poly(dt)、poly(dg)和poly(dc)作为捕获接头片段。另取2组反转录结束后的细胞样本中加入dna连接酶、特定的捕获接头片段以及反应缓冲液,在37℃孵育30分钟,在cdna尾端加上特定的捕获接头片段。另取1组固定结束后的细胞样本,加含反转录酶(moloney murine leukemia virus,)、反转录随机引物、dntps、反转录反应缓冲液、10%tritonx-10的反转录-通透反应试剂进行反转录反应,反转录结束后即在cdna尾端加上三个的dc作为捕获接头片段。加完捕获接头的细胞样本根据上文实施例4、5、6中描述的方法进行细胞分隔、合成cdna第二条链、构建文库。结果显示不同捕获接头片段的细胞样本的ct值范围为12~13,说明本发明方法中使用不同捕获接头片段均可在细胞中捕获较多cdna(参见图12a),凝胶电泳结果显示得到弥散状条带,条带大小分布主要为200~500bp(参见图12b),说明本发明方法中使用不同捕获接头片段在细胞中捕获的cdna捕获率高、覆盖范围均较广,均可用于后续高通量测序。
93.实施例8:对小鼠脑样本进行测序准备(不同反转录和细胞分隔体系的对比)
94.方案一:小鼠断颈处死后分离脑组织,使用液氮将脑组织粉碎,使用裂解液将组织粉末解离成单细胞核。取约600万小鼠脑细胞核样本,加入pbs缓冲液洗三次,加入1%多聚甲醛,置于4℃固定过夜。固定结束后将细胞核样本用pbst缓冲液洗三次。取100万小鼠细胞核参照上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库,其中反转录步骤中使用不携带标签序列的反转录随机引物,细胞分隔时形成单个腔室包含一个单细胞的细胞分隔。
95.方案二:取500万小鼠细胞核参考上文实施例1中描述的方法进行反转录、加捕获接头、细胞分隔、合成cdna第二条链、构建文库,其中反转录过程在96孔板中进行,与方案一相比,每孔反应体系缩小为10ul,每孔使用的引物为携带标签序列的反转录引物,细胞分隔时使用10倍浓度的细胞样品(2000~4000个/ul),其他试剂和微球浓度不变,细胞分隔后至少部分单个腔室中包含多个单细胞,同时空液滴数量降低。
96.根据附图可知,在收集相同液滴数量条件下,方案二获得更多细胞数量,同时检测到的基因数量和分群情况和方案一具有高度的一致性(图13)。这说明本发明方法中通过在反转录过程中添加标签序列不但可以增加单次测量时的细胞通量,还可以增加细胞捕获的效率,降低成本。
97.以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应当了解,在本发明不受上述实施例的限制,上述实施例和说明书中的描述只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1