一种高效人类及哺乳动物细胞附着体表达载体、构建方法和应用
1.本发明属于基因工程及基因治疗技术领域,具体涉及一种高效人类及哺乳动物细胞附着体表达载体,同时还涉及该附着体表达载体的构建方法和应用。
背景技术:
2.基因治疗为现代医学的许多领域带来了新的治疗可能性。基因治疗是指将正常基因或治疗性dna序列以特定方式导入靶细胞中,以纠正缺陷基因,从而治愈疾病。自20世纪90年代初首次成功的基因治疗临床试验以来,已经完成了数千项基因治疗临床试验。基因治疗的成功在很大程度上取决于选择合适的基因传递载体,主要包括病毒载体和质粒载体。虽然病毒载体具有基因转移效率高的优势,但病毒蛋白的产生可能会对宿主细胞造成病理损伤。质粒载体不产生病毒蛋白,但它们的转染效率比较低。根据载体进入宿主细胞后的存在状态,可将载体分为整合载体和附着体载体。将外源基因整合到宿主细胞染色体上,很容易诱发插入突变,安全性较低。与整合载体不同,附着体载体不会整合到基因组中,而是附着在染色体上,它降低了插入突变的风险,具有更高的安全性。
3.申请公布号为cn102703503a的中国发明专利申请公开了一种人类和哺乳动物细胞附着表达载体及其应用,其将2200 bp人β-干扰素核基质结合区(matrix attachment region,mar)序列剪切并拼接成367 bp大小的mar特征性序列构建非病毒附着体载体pegfp-c1-m,pegfp-c1-mar是一种新型的哺乳动物细胞表达载体,能附着而非整合到中国仓鼠卵巢(chinese hamster ovary, cho)细胞染色体上,克服传统载体整合效应带来的副作用,并能驱动转基因表达。但在研究中发现载体本身的cmv启动子驱动外源基因表达拷贝数低、基因表达水平低且不稳定。为了解决该技术问题,申请公布号为cn105802997a的中国发明专利将pegfp-c1-mar的cmv启动子替换为人类延伸因子1α(elongation factor-1α, ef-1α)启动子构建peme附着体载体,发现ef-1α启动子可以驱动非病毒附着体载体高效稳定表达,尽管筛选了较优的启动子,然而载体的转基因表达仍然存在不稳定性和低拷贝数(6-12个拷贝/细胞)的问题。
4.调控元件是调控转基因表达而不编码任何蛋白质的dna序列,研究发现内含子、ucoe、star 40、mar等调控元件具有提高整合载体转基因表达水平的作用,但其作用与调控元件的种类、方向及位置有关。如何组合使用这些调控元件,选择合适的方向和位置,进一步提高附着表达载体转基因表达的稳定性和拷贝数,成为了目前需要解决的技术问题。
技术实现要素:
5.为了克服现有技术的缺陷,本发明的目的之一在于提供高效人类及哺乳动物细胞附着体表达载体,该表达载体相比现有同类型载体,具有更高的稳定性,进一步提升目的基因表达量。
6.本发明的目的之二在于提供高效人类及哺乳动物细胞附着体表达载体的构建方
法。
7.本发明的目的之三在于提供高效人类及哺乳动物细胞附着体表达载体的应用。
8.为了实现上述发明目的,本发明采用的技术方案如下:一种高效人类及哺乳动物细胞附着体表达载体,包括启动子序列,启动子序列上游反向插入第一核基质结合区序列,启动子序列下游正向插入内含子序列,内含子序列下游正向插入第二核基质结合区序列。
9.作为优选的,所述启动子序列为ef-1α序列,如seq id no:1所示。
10.进一步的,所述第一核基质结合区序列为mar x-29序列,如seq id no:2所示。
11.更进一步,所述内含子序列为hcmv内含子序列,如seq id no:3所示。
12.可选的,所述内含子序列与第二核基质结合区序列之间还包括报告基因序列;所述第二核基质结合区序列为mar特征性序列,如seq id no:4所示。在本发明的一些实施例中,所述报告基因序列为egfp。
13.可选的,所述出发载体可以使用任何人或哺乳动物细胞表达载体,作为举例说明,在本发明的一些实施例中,所述出发载体为pegfp-c1,或者按照专利cn102703503a方法构建的pegfp-c1-mar,或者按照专利cn105802997a构建的peme。
14.上述高效人类及哺乳动物细胞附着体表达载体可按照基因工程领域的常规方法构建,作为举例说明,在本发明的一些实施例中其构建方法包括以下步骤:1)人工合成启动子序列,将启动子序列和出发载体双酶切后连接、转化与鉴定,构建形成中间载体1;2)人工合成核基质结合区序列,将核基质结合区序列和中间载体1双酶切后连接、转化与鉴定,构建形成中间载体2;3)人工合成内含子序列,将内含子序列和中间载体2双酶切后连接、转化与鉴定,构建形成所述载体。
15.上述高效人类及哺乳动物细胞附着体表达载体作为外源基因传递载体方面的应用,将外源基因克隆至附着体表达载体形成外源基因表达载体,外源基因表达载体转染宿主细胞表达目的蛋白。
16.作为举例说明,所述外源基因为hsv-tk基因(genbank登录号:jx392980.1)。
17.作为举例说明,所述宿主细胞为人结肠癌细胞hct116。
18.本发明的有益效果:本发明将核基质结合区序列、内含子序列以及启动子序列组合作为人或哺乳动物附着体表达载体的元件,在启动子上游反向插入第一核基质结合区序列,启动子下游正向插入内含子序列,内含子序列下游正向插入第二核基质结合序列,并进一步优选启动子序列为ef-1α序列、第一核基质结合区序列为mar x-29、内含子序列为hcmv内含子序列、第二核基质结合区序列为mar特征性序列,提高附着体表达载体持续、高效和稳定表达外源目的基因。并通过实验验证,使用本发明构建的表达载体,宿主细胞采用cho细胞或人结肠癌细胞hct116,能显著提高转基因表达水平与表达稳定性,表达的目的蛋白具有生物学功能。
附图说明
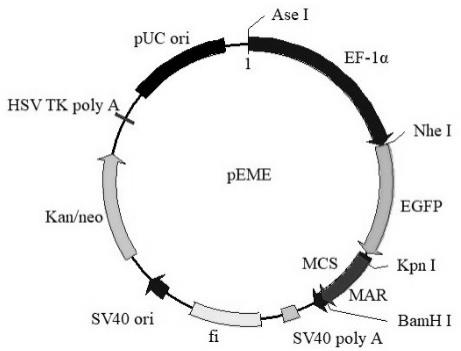
19.图1为重组质粒peme的结构示意图;
buffer 1μl,nhe
ꢀⅰꢀ
酶(10u/μl)0.5μl,补三蒸水至20μl,充分混匀,37℃水浴酶切6h,酶切结束后,于1.5%琼脂糖凝胶进行电泳,凝胶回收酶切后的片段再进行eco 47
ꢀⅲꢀ
酶切;回收的peme(1μg/μl)10μl,10
×ꢀ
o buffer 2μl,eco 47
ꢀⅲꢀ
酶(10u/μl)0.5μl,补三蒸水至20μl,充分混匀,37℃水浴酶切6h;s3:连接线性peme质粒(1μg/μl)5μl,酶切后的正向hcmv内含子片段(1μg/μl)1μl,t4 ligase buffer 1μl,t4连接酶(350u/μl)0.5μl,补三蒸水至20μl,充分混匀,16℃过夜连接;s4:转化与鉴定无菌状态下取200μl新鲜制备的e.coli jm109感受态细菌于灭菌的1.5ml ep管中,加入10μl上述连接好的反应溶液进行转化,接下来接种在含卡那霉素的琼脂平板上, 37℃培养过夜,从培养的平板上挑取阳性转化子放置于3ml lk液体培养基的试管中, 37℃摇床培养过夜;提取重组质粒,并进行重组质粒的双酶切验证,选取酶切验证正确的质粒,进行测序验证,将目的基因序列完全正确的载体命名为peme-hcmvi(f)(如图2所示);2、构建pmar(r)
ꢀ‑
hcmv(f)s1:参照mar x-29 dna片段序列(genbank:ef694970.1),合成其反向(revers, r)序列,基因合成由通用生物系统(安徽)有限公司完成;s2:采用无缝克隆的方法,将mar x-29反向片段,插入到peme-hcmvi(f)载体(图2)ef-1α启动子表达盒上游,将构建好的重组载体转化感受态细胞e.coli jm 109,然后接种在含卡那霉素的培养基中37℃过夜,挑取单菌落继续培养在含卡那霉素的培养基中,37℃、300r/min摇菌过夜;提取重组质粒;取10μl重组dna质粒进行kpn i/apa i双酶切鉴定,选取酶切验证正确的质粒,进行测序验证,将测序正确的载体命名为pmar (r)
ꢀ‑
hcmv(f)(图3所示)。
23.对比例1本对比例按照建上述实施例1构建peme-hcmvi(f)的同样方法,人工合成hcmvi反向序列,构建peme-hcmvi(r),如图4所示。
24.对比例2本对比例构建peme-mar (f):采用无缝克隆的方法,将mar x-29正向片段插入到peme载体(图1)ef-1α启动子表达盒上游插入mar x-29的附着体表达载体。将构建好的重组载体转化感受态细胞e.coli jm 109,然后接种在含卡那霉素的培养基中37℃过夜,挑取单菌落继续培养在含卡那霉素的培养基中,37℃、300r/min摇菌过夜;采用sds碱裂解法提取重组质粒;取10μl重组dna质粒进行酶切鉴定,用nhe i/kpn i双酶切,选取酶切正确的质粒,进行测序验证,验证正确的载体命名为peme-mar (f),如图5所示。
25.对比例3本对比例构建peme-mar (r):采用无缝克隆的方法,将mar x-29反向片段插入到peme载体(图1)ef-1α启动子表达盒上游构建含mar x-29的附着体表达载体。将构建好的重组载体转化感受态细胞e.coli jm 109,然后接种在含卡那霉素的培养基中37℃过夜,挑取单菌落继续培养在含卡那霉素
的培养基中,37℃、300r/min摇菌过夜;采用sds碱裂解法提取重组质粒;取10μl重组dna质粒进行酶切鉴定,用spe i/apa i双酶切,选取酶切正确的质粒,进行测序验证,验证正确的载体命名为peme-mar (r),如图6所示。
26.试验例1试验方法:将出发载体、实施例1、对比例1~3构建的表达载体分别转染cho细胞后检测报告基团egfp表达情况:选择生长状态良好的cho-k1细胞接种到6孔培养板上,待铺板密度达到约80%进行转染。根据实验设计转染共分为6组:未转染组,转染peme、peme-mar (f)、peme-mar (r)、peme-hcmvi(f)、 peme-hcmvi(r)、pmar (r)
ꢀ‑
hcmv(f)载体组。取上述载体4μg加到250μl不含血清和抗生素的dmem-f2培养基中,轻轻混匀,10μl脂质体2000试剂稀释于250μl不含血清和抗生素的dmem-f2培养基中,轻轻混匀,室温放置5min;脂质体2000稀释液滴加到质粒dna稀释液中,一边滴加一边混匀,室温孵育20min,接着将脂质体2000/dna复合物加到每个空中并轻轻摇动使其混合,放入5%co2细胞培养箱中,37℃培养6h后将无血清培养基换为完全的dmem培养基。转染48h后,流式细胞仪检测各组细胞egfp的平均荧光强度(mean florescence intensity,mfi)。
27.试验结果:瞬时表达:与对照组peme相比,除了peme-hcmvi(r),其余重组载体均提高了egfp的瞬时表达水平,将对照组egfp的mfi设置为1,peme-mar (f)、peme-mar (r)和peme-hcmvi(f)组分别提高了1.15,1.50和1.34倍,如图7所示;稳定细胞株的筛选:细胞转染后48h,用终浓度为800μg/ml的g418培养液加压筛选,每3d更换培养液,7-10天未转染组细胞死亡后,g418培养液改为维持浓度400μg/ml持续筛选2周,待稳定转化的细胞集落形成后,继续培养,待细胞密度达80%~90%时,收集细胞并用流式细胞术检测稳定egfp的表达量。对照组peme的egfp表达水平为3.05
×
105,peme mar (r)组为4.88
×
105,peme-hcmv(f)组4.27
×
105,与瞬时表达相似,与对照组peme相比,peme-mar (f)、peme-mar (r)和peme-hcmvi(f)组egfp的稳定表达水平分别提高了1.29,1.67和1.46倍,而peme-hcmvi(r)组未提高egfp的表达水平,如图8所示;pmar (r)
ꢀ‑
hcmv(f)为9.49
×
105。与peme组相比,pmar (r)
ꢀ‑
hcmv(f)组提高了egfp的表达水平达3.11倍,与peme mar (r)组相比提高了1.95倍,与peme-hcmv(f)组相比提高了2.22倍,如图9所示,说明在启动子上游反向插入mar x-29与启动子下游正向插入hcmv内含子的组合可以显著提高转基因的稳定表达水平。
28.试验例2试验方法:使用实施例1构建的表达载体构建含有目的基因hsv-tk的附着表达系统:1 hsv-tk序列的合成hsv-tk序列(genbank:jx392980.1),由通用生物系统(安徽)有限公司合成,为实现定向克隆,在序列的两端分别引入nhe
ꢀⅰ
、kpn i酶切位点。
29.2 构建含hsv-tk目的基因的表达载体用nhe i/kpn i双酶切合成的hsv-tk序列,同时用nhe i/kpn i双酶切pmar(r)-hcmv(f)质粒,采用常规的酶切方法,37℃酶切6h;琼脂糖凝胶电泳鉴定酶切结果,凝胶回收
hsv-tk序列片段和pmar(r)-hcmv(f)线形质粒;酶切回收后的pmar(r)-hcmv(f)线性质粒dna及hsv-tk片段按摩尔比 5:1的比例进行16℃过夜连接;然后将连接产物转化到e.colijm109菌株感受态细胞悬液中,将菌液接种在含有卡那霉素的lb平板上,37℃培养过夜;挑取单菌落进行培养,并进行重组质粒的提取;提取的质粒进行nhe i/kpn i双酶切验证,选取酶切验证正确的质粒进行测序验证,将测序正确的载体命名为pmar(r)-hcmv(f)-hsv-tk;3 载体转染hct16细胞表达系统的建立生长状态良好的hct16细胞接种到6孔培养板上,待铺板密度达到约80%-90%时进行转染。具体操作步骤如下:将10μl lipofectamine2000添加到250μl无血清rpmi-1640培养基中,同时将250μl无血清的rpmi-1640培养基与4μg表达质粒混合;接下来脂质体2000稀释液滴加到质粒dna稀释液中,一边滴加一边混匀,室温孵育20min后将脂质体2000/dna复合物的混合液轻轻滴入孔中;放入5%co2的细胞培养箱中37℃培养6h后,将无血清的rpmi-1640培养基更换成rpmi-1640完全培养基,放入细胞培养箱内继续培养;4 目的基因hsv-tk的表达分析转染48h后,pbs洗涤培养细胞,胰蛋白酶进行消化,一部分细胞加入rpmi-1640细胞培养基继续培养;另一部分细胞收集到1.5 ml ep管中以1000 rpm离心5 min以收集细胞沉淀;向细胞沉淀中加入ripa裂解液,13000 rpm离心15 min收集上清;western bolt检测hsv-tk瞬时表达。
30.转染48h后进行稳定株的筛选:细胞在含有800 ug/ml g418)完全培养基中继续培养,约1 w后空白对照细胞死亡,更换为400 ug/ml g418的rpmi-1640完全培养基进一步培养,稳定筛选约5 w后获得稳定细胞株,收集稳定细胞株沉淀,加入ripa裂解液,13000 rpm离心15 min收集细胞上清,western bolt检测hsv-tk稳定表达。western blot结果发现,在瞬时和稳定筛选的hct116细胞内都可以检测到hsv-tk的表达,经灰度分析稳定表达水平比瞬时表达提高了2.15倍,如图10所示;5 hsv-tk蛋白的功能验证前体药物丙氧鸟苷(ganciclovir, gcv)是一种核苷酸类似物,在hsv-tk的催化作用下形成一磷酸丙氧鸟苷,然后再转化为三磷酸丙氧鸟苷,干扰细胞分裂时dna的合成,最终导致细胞凋亡。应用gcv后不但转基因的肿瘤细胞被杀灭,周围大量未转染的细胞也被杀死,这种现象就称为旁观者效应。旁观者效应可用于鉴定目的基因hsv-tk的功能,低于50% hsv-tk阳性细胞可以杀伤60% 以上的细胞认为自杀基因有效。细胞旁效应的具体操作步骤如下:将稳定转染的hct116细胞株和未转染的细胞在不同浓度下混合(0∶10、1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1、10∶0)并接种于96孔板中,每孔接种5000个细胞,在37℃和5% co2的培养箱培养中培养;细胞贴壁后,培养基替换为含gcv 80 ug/ml的培养基;培养48 h后,20 ul mtt溶液(5 mg/ml)加入每个孔中,锡箔包裹继续培养4 h;弃去孔内培养液,150 ul dmso加入到每个孔中,并在摇床上低速摇动10 min,以完全溶解晶体;在酶联免疫检测仪测量各孔的吸光度值,计算细胞生长抑制率。结果发现,当稳定转染的hct116细胞株占比为10% 时,细胞抑制率达到33.25%,当占比达到40%时,细胞抑制率为75.87%,当占比达到50%时,细胞抑制率为82.67%,如图11所示,表明少量稳定转染的hct116细胞株可引起大多数细胞凋亡,即旁观者效应效果明显,自杀基因有功能。
31.最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1