苯并杂环类化合物及其制备方法、药物组合物和应用
1.本发明涉及一种苯并杂环类化合物及其制备方法、药物组合物和应用,具体涉及一种对sting蛋白具有激动作用的苯并杂环类化合物及其制备方法、药物组合物和应用。
背景技术:
2.人类的免疫系统包括先天性免疫与适应性免疫,它通过一系列免疫相关的共刺激与共抑制信号分子的精确调控来实现。其中,先天性免疫反应在宿主有效防御微生物等相关病原入侵中发挥重要作用。入侵的病原体被宿主细胞中的模式识别受体(prrs)识别,之后启动一系列信号事件,促使炎性细胞因子、i型干扰素及下游其他抗病毒蛋白的表达。细胞中免疫信号通路的异常将导致癌症在内的多种疾病的发生。
3.干扰素基因刺激物(stimulator of interferon genes,sting)作为一种环状二核苷酸(cdns)的prrs,在激活宿主免疫反应,对抗病毒感染、介导炎症及肿瘤免疫治疗中发挥着重要的作用。sting可被多种环二核苷酸(cyclic dinucleotide,cdn)激活,其中cgas(cyclic gmp-amp synthase,cgas)产生的内源性(2
′
,3
′‑
cyclic guanosine adenosine monophosphatec,gamp)是其亲和力最高的配体。cgas是胞质内的一种双链dna(double stranded dna,dsdna)识别受体,可识别胞质中各种来源dsdna。cgas与dsdna结合后会催化amp与gmp生成第二信使cgamp。cgas、cgamp和sting构成了哺乳动物胞质中最主要的dna感知途径。cgas-sting信号通路在机体应对病毒和细菌感染、肿瘤侵袭的先天免疫过程中不可或缺,激活sting有望对上述疾病的治愈产生重要的作用。
4.近年来,cgas-sting通路作为宿主介导对dna核酸免疫反应的主要途径之一,已逐渐成为抗肿瘤免疫治疗中的新的候选者。目前sting靶点还没有新药小分子上市,已进入临床的sting激动剂大多是cdn类化合物,化学结构类型比较有限。
技术实现要素:
5.发明目的:针对现有化合物存在的结构单一、疗效有限等问题,本发明旨在提供一种具有sting激动活性的苯并杂环类化合物及其制备方法、药物组合物和应用。
6.技术方案:作为本发明涉及的第一方面,本发明的苯并杂环类化合物具有式(i)的结构,还包含其药学上可接受的盐:
[0007][0008]
其中:
[0009]
x代表碳或氮;
[0010]
m=0、1或2;
[0011]
n=0、1或2;
[0012]
p=0、1或2;
[0013]
a代表苯基或苯并芳杂环基,所述苯环或苯并芳杂环被一个或多个以下基团取代:氢、卤素、氰基、氨基、羟基、三氟甲基、三氟甲氧基、氨基甲酰基、c1~c4烷基或c
1-c4烷氧基;
[0014]
b代表
[0015]
r1或r2代表氢、氘、卤素、氰基、三氟甲基、c1~c3烷基或3~6元环烷基;
[0016]
r3或r4代表c1~c3烷基或3~6元环烷基;
[0017]
r5代表c1~c3烷基或3~6元环烷基;
[0018]
r6代表苯基或芳杂环基,所述苯基或芳杂环基被一个或多个以下基团取代:氢、卤素、氰基、氨基、羟基、三氟甲基、三氟甲氧基、甲酰基、c1~c4烷基或c1~c4烷氧基。
[0019]
优选,上述结构中:
[0020]
m=0或1;
[0021]
n=0或1;
[0022]
p=0或1;
[0023]
a选自苯基或取代的苯并芳杂环基,所述苯基或苯并芳杂环基被一个或多个以下基团取代:氢、卤素、氰基、氨基、羟基、三氟甲基、三氟甲氧基或氨基甲酰基;
[0024]
b代表
[0025]
r1或r2代表氢、氘、卤素、氰基或三氟甲基;
[0026]
r3或r4代表氢或c1~c3的烷基;
[0027]
r5选自c1~c3烷基;
[0028]
r6代表苯基或芳杂环基,所述苯基或芳杂环基被一个或多个以下基团取代:氢、卤素或三氟甲基。
[0029]
进一步优选,上述结构中:
[0030]
a代表其中r7代表一个或多个氢、卤素、甲基、氨基甲酰基。
[0031]
进一步优选,上述结构中:
[0032]
m=0,p=1。
[0033]
进一步优选,上述结构中:
[0034]
r1或r2代表氘或氟。
[0035]
最优选,上述苯并杂环类化合物为以下任一化合物:
[0036][0037]
[0038]
上述苯并杂环类化合物的药学上可接受的盐为所述苯并杂环类化合物与酸形成的盐,所述酸为盐酸、氢溴酸、硫酸、磷酸、碳酸、甲磺酸、苯磺酸、对甲苯磺酸、萘磺酸、柠檬酸、酒石酸、乳酸、丙酮酸、乙酸、马来酸、琥珀酸、富马酸、水杨酸、苯基乙酸、杏仁酸或阿魏酸。
[0039]
作为本发明涉及的第二方面,上述苯并杂环类化合物由以下任一方法制备得到:
[0040]
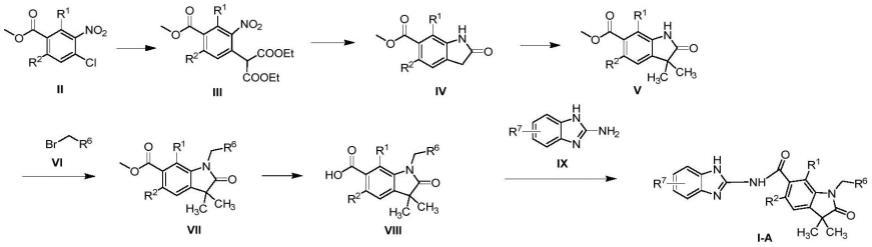
(1)当n=0、m=0、p=1、a为x为碳、b为r1或r2为氢、氟或氘、r3为甲基、r4为甲基时,目标化合物i-a的制备方法如下:
[0041][0042][0043]
其中,r6、r7的定义同前。
[0044]
具体地,由化合物ii制备化合物iii,是将丙二酸二乙酯溶于溶剂中,0℃下加入氢化钠,1h后加入ii移至室温条件下得到。所用溶剂选自甲苯、dmf、dmac、乙二醇二甲醚、乙二醇单甲醚、1,4-二氧六环、thf、甲醇、乙醇、乙腈、丙酮等,优选dmf。
[0045]
由化合物iii制备化合物iv,是将iii溶于溶剂中,加入铁粉和氯化铵后加热,还原硝基得到,所用溶剂选自二氯甲烷、二氯乙烷、三氯甲烷、四氯化碳、thf、甲醇、甲苯、乙醇、乙腈、dmf或上述任意两种或三种溶剂组成的混合溶剂,优选乙醇和水的混合体系。
[0046]
由化合物iv制备化合物v,是将iv溶于溶剂中,0℃下加入氢化钠,1h后加入碘甲烷移至室温条件下得到。所用溶剂选自甲苯、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、乙二醇二甲醚、乙二醇单甲醚、1,4-二氧六环、四氢呋喃(thf)、甲醇、乙醇、乙腈、丙酮等,优选dmf。
[0047]
由化合物v制备化合物vii,是将v溶于溶剂中,0℃下加入氢化钠,1h加入vi,加毕移至室温条件下得到。所用溶剂选自甲苯、dmf、dmac、乙二醇二甲醚、乙二醇单甲醚、1,4-二氧六环、thf、甲醇、乙醇、乙腈、丙酮等,优选dmf。
[0048]
由化合物vii制备化合物viii,是将vii溶于溶剂中,加入碱的水溶液,室温条件下反应得到。所用的碱选自氢氧化钠、氢氧化钾、氢氧化锂等,优选氢氧化锂。所用的溶剂选自
甲苯、dmf、dmac、乙二醇二甲醚、乙二醇单甲醚、1,4-二氧六环、thf、甲醇、乙醇、乙腈、丙酮、水和任意两种溶剂组成的混和溶剂等,优选thf和水的混和溶液。
[0049]
由化合物viii制备化合物i-a,是将viii溶于溶剂中,加入缩合剂,再加入碱和化合物ix进行缩合反应得到。所用溶剂选自二氯甲烷、thf、dmf、1,4-二氧六环、乙腈等,优选dmf。所用缩合剂选自n,n'-羰基二咪唑(cdi)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)、n,n'-二环己基碳二亚胺(dcc)、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)、1-羟基苯并三唑(hobt)等,优选hatu。所用碱选自三乙胺、碳酸钠、碳酸钾、n,n-二异丙基乙胺,优选n,n-二异丙基乙胺。
[0050]
(2)当n=1、m=0、p=1、a为x为碳、b为r1或r2为氢、氟或氘、r3为甲基、r4为甲基时,目标化合物i-b的制备方法如下:
[0051][0052]
其中,r6、r7的定义同前。
[0053]
具体地,由化合物viii制备化合物i-b,是将viii溶于溶剂中,加入缩合剂,再加入碱和化合物x进行缩合反应得到。所用溶剂选自二氯甲烷、thf、dmf、1,4-二氧六环、乙腈等,优选dmf。所用缩合剂选自cdi、edci、dcc、dic、hatu、hobt等,优选hatu。所用碱选自三乙胺、碳酸钠、碳酸钾、n,n-二异丙基乙胺,优选n,n-二异丙基乙胺。
[0054]
将相应的酸与以上方法制备得到的化合物(i)成盐,即得所述苯并杂环类化合物的药学上可接受的盐。
[0055]
作为本发明涉及的第三方面,上述苯并杂环类化合物与药学上可接受的载体形成药物组合物。具体地,上述苯并杂环类化合物可以添加药学上可接受的载体制成常见的药用制剂,如片剂、胶囊、糖浆、悬浮剂或注射剂,制剂可以加入香料、甜味剂、液体/固体填料、稀释剂等常用药用辅料。
[0056]
作为本发明涉及的第四方面,上述苯并杂环类化合物及其药物组合物可制备为sting激动剂药物,具体为抗肿瘤的药物。
[0057]
有益效果:与现有技术相比,本发明具有如下显著优点:
[0058]
(1)该类化合物可对sting蛋白产生显著的激动作用,有效促进干扰素β分泌;
[0059]
(2)该类化合物及其药物组合物在细胞水平上具有优异的sting激动作用,可用于肿瘤的免疫治疗;
[0060]
(3)化合物制备便捷、易于操作。
具体实施方式
[0061]
下面结合实施例对本发明的技术方案作进一步说明。
[0062]
实施例1:2-(1-(3-氟苄基)-3,3-二甲基-2-氧吲哚啉-6-羧酰氨基)-1h-苯并[d]咪唑-5-羧酰胺(i-a-1)
[0063]
2-(4-(甲氧基羰基)-2-硝基苯基)丙二酸二乙酯(iii)的合成
[0064]
在250ml茄形瓶中,将丙二酸二乙酯(7.43g,46.38mmol)溶于125ml dmf中,置于冰浴中。溶液澄清,无色透明。10min内分批次加入60%nah(2.78g,69.57mmol),可见有气泡放出,溶液变浑浊。冰浴中搅拌15min后,向反应液中加入4-氯-3-硝基苯甲酸甲酯(5.00g,23.19mmol),并将其移至室温下反应1h,反应液颜色逐渐变为深红色。tlc(v石油醚∶v乙酸乙酯=2∶1)监测原料反应完毕,将反应液倒入冰中,并用2mol/l稀盐酸调ph至2~3,乙酸乙酯(100ml
×
3)萃取,收集有机相,饱和氯化钠溶液(100ml
×
3)洗涤,无水硫酸钠干燥。硅胶柱层析纯化(v石油醚∶v乙酸乙酯=10∶1)得白色固体4.58g。产率:58%。m.p.200-202℃。1h nmr(300mhz,dmso-d6)δ(ppm):8.60(d,j=2.0hz,1h),8.37(dd,j=8.1,2.0hz,1h),7.79(d,j=7.9hz,1h),5.62(s,1h),4.25(q,j=7.1hz,4h),3.98(s,3h),1.25(t,j=7.1hz,6h).
[0065]
2-氧代吲哚啉-6-甲酸甲酯(iv-1)的合成
[0066]
在250ml的三颈瓶中,将iii(3.42g,10.08mmol)溶于80ml乙酸中,溶液澄清透明淡黄色。机械搅拌下加入还原铁粉(2.41g,43.18mmol),升温至120℃下回流搅拌。反应过程中可见溶液变黑而后生成大量灰白色固体。tlc(v石油醚∶v乙酸乙酯=2∶1)监测反应完全。反应结束后硅藻土抽滤除去铁粉,收集滤液,减压浓缩除去大部分乙酸,饱和nahco3调ph至中性,乙酸乙酯萃取,饱和氯化钠溶液洗有机相,无水硫酸钠干燥后,柱层析(v石油醚∶v乙酸乙酯=200∶1)纯化,得灰白色固体1.50g。产率:78%;m.p.202-204℃。1h nmr(300mhz,chloroform-d)δ(ppm):8.21(s,1h),7.78(dd,j=7.7,1.5hz,1h),7.56(d,j=1.5hz,1h),7.32(d,j=7.8hz,1h),3.94(s,3h),3.62(s,2h).
[0067]
3,3-二甲基-2-氧代吲哚啉-6-羧酸甲酯(v-1)的合成
[0068]
在100ml的茄形瓶中,将化合物iv-1(1.12g,5.86mmol)加入30ml dmf中溶解,溶液澄清透明淡黄色。将反应液置于冰浴中,分批加入60%nah(0.49g,12.31mmol)并搅拌10min,反应液有气泡放出,颜色变为橘黄色。加入ch3i(1.75g,12.31mmol),可见溶液变浑浊。tlc(v石油醚∶v乙酸乙酯=1∶1)检测反应结束。将反应液倒入冰中,乙酸乙酯(30ml
×
3)萃取,饱和氯化钠溶液(50ml
×
3)洗涤,无水硫酸钠干燥。硅胶柱层析(v石油醚∶v乙酸乙酯=5∶1)纯化,得白色固体0.81g。产率:63%;m.p.201-203℃。1h nmr(300mhz,chloroform-d)δ(ppm):9.28(s,1h),7.82(dd,j=7.7,1.5hz,1h),7.68(d,j=1.5hz,1h),7.29(d,j=7.8hz,1h),3.96(s,3h),1.47(s,6h).
[0069]
1-(3-氟苄基)-3,3-二甲基-2-氧吲哚啉-6-甲酸甲酯(vii-1)的合成
[0070]
在50ml茄形瓶中,将化合物xvi-1(0.70g,3.19mmol)溶于15ml dmf中,溶液澄清透明淡黄色。将反应液置于冰浴,分批次加入60%nah(0.18g,4.47mmol),有气泡放出,溶液变黄,有絮状物产生。搅拌10min后加入3-氟溴苄(18)(0.66g,3.51mmol),撤去冰浴,室温下反应约1h。tlc(v石油醚∶v乙酸乙酯=4∶1)监测。结束后将反应液倒入冰中淬灭,2mol/l盐酸调ph至2~3,乙酸乙酯(20ml
×
3)萃取,饱和氯化钠溶液(40ml
×
3)洗涤,无水硫酸钠干燥。硅胶柱层析(v石油醚∶v乙酸乙酯=15∶1)纯化,得白色固体1.01g。产率:97.1%;m.p.125-128℃。1h nmr(300mhz,chloroform-d)δ(ppm):7.78(dd,j=7.7,1.4hz,1h),7.36(d,j=1.5hz,1h),7.34
–
7.27(m,2h),7.07(d,j=8.0hz,1h),7.02
–
6.90(m,2h),4.94(s,2h),3.88(s,3h),1.46(s,6h).
[0071]
1-(3-氟苄基)-3,3-二甲基-2-氧吲哚啉-6-羧酸(viii-1)的合成
[0072]
在50ml的茄形瓶中,将化合物xvii-1(0.20g,0.61mmol)溶于15ml thf与7.5ml h2o的混合溶剂中,溶液澄清,无色透明。加入lioh
.
h2o(0.13g,3.05mmol),室温下搅拌过夜。tlc(v石油醚∶v乙酸乙酯=4∶1加1滴醋酸)监测反应完全。结束后向反应液中加30ml水,调ph至2~3,乙酸乙酯(20ml
×
3)萃取,无水硫酸钠干燥,硅胶柱层析(纯dcm)纯化,得白色固体0.17g。产率:88%;m.p.200-202℃。1h nmr(300mhz,dmso-d6)δ(ppm):12.97(s,1h),7.69(dd,j=7.7,1.4hz),7.53(d,j=7.7hz,1h),7.45
–
7.34(m,2h),7.17
–
7.00(m,3h),5.01(s,2h),1.38(s,6h).
[0073]
2-(1-(3-氟苄基)-3,3-二甲基-2-氧吲哚啉-6-羧酰氨基)-1h-苯并[d]咪唑-5-羧酰氨(i-a-1)的合成
[0074]
在25ml的茄形瓶中,将化合物viii-1(0.25g,0.80mmol)溶于5ml茄形瓶中,依次加入hatu(0.37g,0.96mmol)、dipea(0.41g,3.18mmol),溶液澄清透明淡黄色。室温下搅拌20min,可见反应液颜色由淡黄色变为深红色。tlc(v石油醚:v乙酸乙酯=1∶1)监测原料全部转化为活性酯后,向反应液中加入ix-1(0.17g,0.96mmol)。室温下搅拌约5h。tlc(v二氯甲烷∶v甲醇=8∶1)监测活性酯反应完毕后,向反应液中加水30ml,乙酸乙酯(20ml
×
3)萃取。饱和氯化钠溶液洗有机相3次。柱层析(v二氯甲烷∶v甲醇=60∶1)纯化,得白色固体0.13g。产率:34%;m.p.>260℃。1h nmr(300mhz,dmso-d6)δ(ppm):12.35(s,2h),8.02(d,j=1.6hz,1h),7.93(s,1h),7.91(dd,j=7.8,1.5hz,1h),7.72(dd,j=8.4,1.6hz,1h),7.67(d,j=1.5hz,1h),7.58(d,j=7.8hz,1h),7.47(d,j=8.5hz,1h),7.44
–
7.37(m,1h),7.23(s,1h),7.20
–
7.08(m,3h),5.05(s,2h),1.40(s,6h).hrms(esi):m/z[m+h]
+
calcd for c
26h23
fn5o3:472.1785;found:472.1783.
[0075]
参照实施例1的制备方法,制备得到以下化合物:
[0076][0077]
实施例2:药理学实验——本发明的化合物诱导thp-1细胞内干扰素β水平活性评价
[0078]
1、实验目的
[0079]
使用human ifn-beta duoset elisa检测试剂盒(r&d systems),检测式(i)化合物诱导thp-1细胞内干扰素β水平。
[0080]
2、实验原理
[0081]
elisa即酶联免疫吸附测定,是一类常用的免疫酶技术。主要方法是将已知的抗原或抗体吸附在固相载体表面,然后利用酶标记(偶联)的抗体或抗原与之孵育,加入显色剂显色,通过酶标仪测定待测物颜色与标准物颜色的差异,绘制酶活曲线,得出待测物浓度。固相载体表面的抗体可捕捉到细胞分泌上清液中的干扰素β,并与之结合,而后通过依次连接检测抗体和hrp,最后加入显色剂显色。该颜色在酶标仪的测定下可反映干扰素β释放的水平。
[0082]
3、实验材料
[0083]
thp-1细胞购自中国科学院细胞库;胎牛血清购自sciencell公司;rpmi-1640培养基和生物用dmso购自江苏凯基生物技术股份有限公司;human ifn-beta duoset elisa试剂盒(dy 814-05)和duoset elisa ancillary reagent kit 2购自r&d systems(dy 008)
公司。
[0084]
4、测试仪器
[0085]
酶标仪(thermo electro co.)
[0086]
5、受试化合物
[0087]
式(i)代表的化合物。
[0088]
6、实验过程
[0089]
细胞培养条件:
[0090]
细胞传代培养,培养条件为含有青霉素(终浓度为100u/ml)、链霉素(终浓度为100μg/ml)、10%fbs的培养基(thp-1:1640)。当thp-1细胞生长至对数生长期时,吹打均匀吸取至15ml离心管,1000rpm离心4min。弃去上清液,加入5ml pbs洗涤细胞后再次离心,重复操作一次,用完全培养基重悬细胞,分皿培养,3天换一次液。
[0091]
实验步骤:
[0092]
(1)酶标板的包被:取human ifn-beta duoset elisa试剂盒(r&dsystems,dy814-05)中所需个数的酶标孔板,按照说明书,capture detection(480μg/ml)用pbs/di水进行120倍稀释,每孔100μl加入酶标板,塑封膜塑封放入摇床(110rpm),室温孵育过夜。
[0093]
(2)细胞铺板:取3~5代对数期生长的thp-1细胞,离心后计数铺板,密度1
×
106个/ml。显微镜下观察细胞形态,无异常则进行给药。将配好的待测化合物及阳性药(cgamp)的储备液(50mm)每孔各1μl分别加入所对应的细胞孔板,并留出一个细胞孔作为阴性对照,显微镜下观察后放入37℃恒温培养箱中孵育6h。
[0094]
(3)封闭:倒出酶标板的液体并于纸上轻轻拍干,wash buffer洗板3次。每孔加入300μl reagent diluent(10*)(pbs/di水稀释成1*),室温孵育1~2h,之后重复洗板操作。
[0095]
(4)上样:吹打吸取细胞培养液,于4℃冷冻离心机离心(3000rpm,5min),吸取上清液,每孔100μl加入酶标板,标准品则按说明书进行稀释,每孔100μl加入酶标板,塑封后于25℃500rpm摇床孵育2h。阴性对照不加化合物,空白对照不含包被抗体。
[0096]
(5)孵二抗:重复(3)中洗板操作,detection antibody(15μg/ml)用reagent diluent(1*)进行60倍稀释,每孔100μl加入酶标板,塑封后于25℃500rpm摇床孵育2h。重复(3)中洗板操作,每孔100μl加入reagent diluent(1*)稀释的hrp,避光静置20min。
[0097]
(6)显色:重复(3)中洗板操作,配成substrate solution(color reagent a:color reagent b=1:1),每孔100μl加入酶标板,避光静置20min。之后每孔加入50μl stop solution(2n h2so4),终止反应。于酶标仪波长450nm下读取吸光度od值,结合干扰素β的标准曲线,利用graphpad prism 8软件进行干扰素释放值计算。
[0098]
活性数据参见表1。
[0099]
干扰素β水平:》100pg/ml(记为:a);10.1~100pg/ml(记为:b)。
[0100]
表1.化合物诱导thp-1细胞内干扰素β水平活性
[0101]
化合物编号干扰素β水平(pg/ml)化合物编号干扰素β水平(pg/ml)cgampai-a-4bi-a-1bi-b-1ai-a-2bi-b-2ai-a-3bi-b-3a
[0102]
由表1可见,本发明的化合物可有效促进干扰素β释放,因此可用于肿瘤的免疫治疗。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1