一种异噁唑啉类衍生物的制备方法
1.本发明涉及农药化学技术领域,尤其涉及一种异噁唑啉类衍生物的制备方法。
背景技术:
2.目前,已超过300种杀虫剂流向市场,每十年都会引入至少一个新的化学结构类别,如有机磷、氨基甲酸酯、有机氯、拟除虫菊酯和新烟碱等。传统化学农药因存在高残留、高抗性、高毒性等问题被限制或者禁止使用。如何开发高活性、高选择性、低风险、无残留的产品已成为绿色农药的发展方向。随着研究人员对化学功能分子、作用机理和抗性机制不断深入研究,以氟雷拉纳(fluralaner)为代表的异噁唑啉(isoxazoline)类杀虫剂被成功研制,2014年已作为杀虫类兽药(商品名bravectotm)上市销售,主要用于动物体内外杀虫;2016年7月美国fda批准了默克公司申请的(氟雷拉纳外用溶液)用于治疗猫和狗身上的跳蚤和扁虱,一次给药有效期长达12周。异噁唑啉类杀虫剂的化学结构新颖、杀虫谱广、作用位点独特,无交叉抗性等特点引起了业内广泛的关注。此类杀虫剂对鳞翅目、半翅目、缨翅目、蜱螨目等害虫具有良好的杀虫效果。其作为非竞争性拮抗剂(ncas)作用于昆虫γ-氨基丁酸(gaba)受体,抑制其氯离子内流,干扰了gaba跨膜信号传递,最终使昆虫神经兴奋不能控制而死亡。异噁唑啉类杀虫剂的作用靶点为nca-ii型,与传统的gaba受体ncas,如有机氯类(林丹、狄氏剂、硫丹)和苯基吡唑类(氟虫腈)的靶点nca-ia型相比,不存在交叉抗性,而且更加环保和安全。国内外制药公司相继对此类化合物开展研究,目前已有6个品种商品化或即将商品化,其中2种为农药杀虫剂,4种为抗寄生虫兽药,成为了杀虫剂研究热点领域之一。
3.fluralaner(氟雷拉纳,开发代号:a1443)是由日产化学在2004年研发合成,其于2005年获批专利wo2005085216,存在r、s构型。研究表明fluralaner主要的杀虫活性组分为s构型。
[0004][0005]
现有氟雷拉纳的众多合成方法中,其根源在于存在不同的方法合成异噁唑啉加甲苯的母环结构。当前所用最为广泛的方法是这两种:[cc+cno]分子间关环法和[ccc+no]分子内关环法。
[0006]
fluralaner[cc+cno]分子间关环法:以1,3-二氯-5-(1-三氟甲基-乙烯基)-苯和4-((羟基亚氨基)甲基)-2-甲基苯甲酸甲酯进行1,3-偶极环加成反应,最后再在碱性条件下与2-氨基-n-(2,2,2-三氟乙基)乙酰胺发生酰胺化反应制备得到氟雷拉纳。其合成路线如下:
(3,5-二氯苯基)-1,1,1-三氟戊-4-烯-2-醇,然后使用臭氧对末端双键进行氧化得到(s)-3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁醛;所得到的醛化合物和(4-(甲氧羰基)-3-甲基苯基)氯化镁氯化锂络合物反应后的产物直接使用dess-martin氧化,得到(s)-4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁酰基)-2-甲基苯甲酸甲酯;随后(s)-4-(3-(3,5-二氯苯基)-4,4,4-三氟-3-羟基丁酰基)-2-甲基苯甲酸甲酯在盐酸羟胺以及phi(oh)ots和p(ome)3作用下关环制备得到(s)-4-(5-(3,5-二氯苯基)-5-(三氟甲基)-4,5-二氢异恶唑-3-基)-2-甲基苯甲酸甲酯;最后,(s)-4-(5-(3,5-二氯苯基)-5-(三氟甲基)-4,5-二氢异恶唑-3-基)-2-甲基苯甲酸甲酯和2-氨基-n-(2,2,2-三氟乙基)乙酰胺缩合实现s构型的氟雷拉纳的制备。其合成路线如下:
[0011][0012]
综上所述,可以看出现有的氟雷拉纳合成路线在根源存在共同点,就是合成异噁唑啉环。但目前的关环前体都含有多个活泼的基团,诸如酯基、酰氨基、羧基等,且关环的试剂都为盐酸羟胺,这些活泼的基团非常易于和盐酸羟胺发生副反应,从而导致整个制备工艺产生杂质,为后续的产品纯化和产业化生产带来瓶颈。因此,开发新的氟雷拉纳的合成方法对于氟雷拉纳的产业化以及降低产业化成本、提高该产品的市场竞争力都非常重要。
[0013]
鉴于此,有必要提供一种新的异噁唑啉类衍生物的制备方法。
技术实现要素:
[0014]
本发明为了解决上述技术问题提供一种异噁唑啉类衍生物的制备方法。本发明以1,3-二氯-5-(1-三氟甲基-乙烯基)-苯和二溴甲醛肟为原料,能够合环形成异恶唑环,中间体的产率高,能够合成得到弗雷拉纳,制备反应稳定。
[0015]
本发明解决上述技术问题的技术方案如下:
[0016]
一种异噁唑啉类衍生物的制备方法,所述异噁唑啉类衍生物为弗雷拉纳,其化学
结构式如通式ⅰ所示:
[0017][0018]
制备所述弗雷拉纳的反应方程式如下:
[0019][0020]
上述反应式的具体反应步骤如下:
[0021]
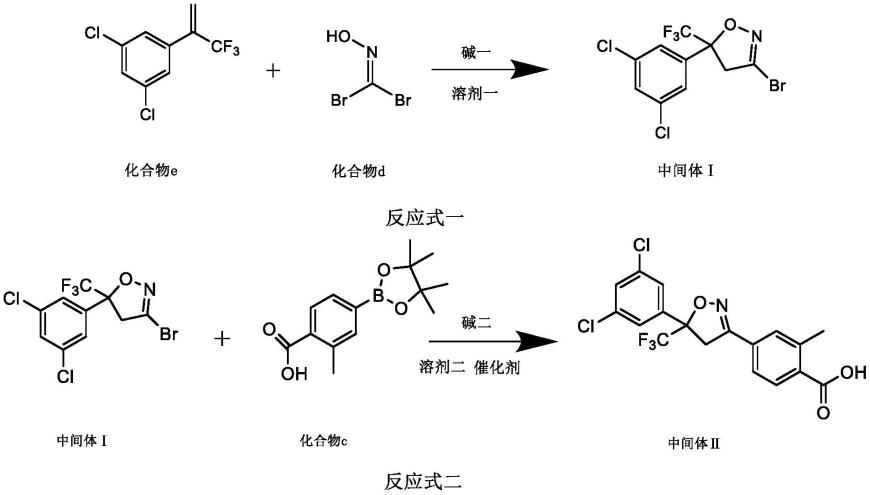
步骤a:如反应式一所示,将化合物e、化合物d和碱一加入到溶剂一中进行关环反应,制得中间体ⅰ;
[0022]
步骤b:如反应式二所示,将中间体ⅰ、化合物c、碱二和催化剂加入到溶剂二中进行suzuki-miyaura反应,制得中间体ⅱ;
[0023]
步骤c:如反应式三所示,将中间体ⅱ、化合物b、碱三和缩合剂加入到溶剂三中进
行酰胺化缩合反应,即得弗雷拉纳。
[0024]
本发明的有益效果是:本发明的制备方法,首先通过合环反应制得含异恶唑环的中间体,然后通过suzuki-miyaura反应和酰胺化缩合反应,制得的弗雷拉纳的产率高,整个制备过程中产生的杂质少,有利于弗雷拉纳的产业化合成生产,降低生产成本,提升弗雷拉纳的市场竞争力。
[0025]
在上述技术方案的基础上,本发明还可以做如下改进。
[0026]
进一步的,步骤a中,所述化合物e、化合物d和碱一的摩尔比为1:(1~2):(3~5)。
[0027]
采用上述进一步方案的有益效果是:在此摩尔比例范围内能够有效的进行关环反应,得到含异恶唑环的中间体i。
[0028]
进一步的,步骤a中,所述溶剂一为乙酸乙酯、四氢呋喃和水中的任意一种或多种;所述碱一为碳酸氢钠和碳酸钠中的任意一种。
[0029]
采用上述进一步方案的有益效果是:采用上述的一,有利于化合物e和d进行关环反应,得到含异恶唑环的中间体i;碱一能够提供碱性环境,有利于减少中间体i的副产物。
[0030]
进一步的,步骤b中,所述中间体ⅰ、化合物c、碱二和催化剂的摩尔比为1:(1~3):(1~3):(0.01~0.03)。
[0031]
采用上述进一步方案的有益效果是:在此摩尔比例范围内能够有效的进行关环反应,得到含异恶唑环的中间体ⅱ。
[0032]
进一步的,步骤b中,所述催化剂为四(三苯基磷)钯和[1,1'-双(二苯基膦基)二茂铁]二氯化钯中的任意一种或两种。
[0033]
采用上述进一步方案的有益效果是:在催化剂作用下,使得suzuki-miyaura反应能够有选择的高效反应。
[0034]
进一步的,步骤b中,所述碱二为无水碳酸钾。
[0035]
采用上述进一步方案的有益效果是:采用污水碳酸钾能够减少suzuki-miyaura反应的副反应的发生。
[0036]
进一步的,步骤b中,所述溶剂二为无水四氢呋喃。
[0037]
采用上述进一步方案的有益效果是:无水四氢呋喃能够提供更好的溶解性,有利于中间体ⅱ的合成。
[0038]
进一步的,步骤c中,所述中间体ⅱ、化合物b、碱三和缩合剂的摩尔比为1:(1~1.5):(1~3):(1~1.5)。
[0039]
采用上述进一步方案的有益效果是:采用上述摩尔比的原料,能够制得弗雷拉纳。
[0040]
进一步的,步骤c中,所述缩合剂为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐。
[0041]
采用上述进一步方案的有益效果是:采用该缩合剂,有利于通过酰胺化缩合反应制得弗雷拉纳。
[0042]
进一步的,步骤c中,所述碱三为4-甲基氨基吡啶和三乙胺中的任意一种;所述溶剂三为二氯甲烷。
[0043]
采用上述进一步方案的有益效果是:采用上述的碱三,能够制得弗雷拉纳;二氯甲烷作为溶剂能够有利于中间体ⅱ和化合物b反应。
具体实施方式
[0044]
以下对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0045]
实施例
[0046]
异噁唑啉类衍生物弗雷拉纳,其化学结构式如下:
[0047][0048]
弗雷拉纳的制备包括如下步骤:
[0049]
1、中间体ⅰ的制备:
[0050][0051]
将化合物e(1.82g,7.56mmol)、化合物d(2.30g,11.34mmol)、碳酸氢钠(2.66g,31.64mmol)和20ml乙酸乙酯加入50ml烧瓶,室温下搅拌12h。tlc(展开剂为纯石油醚)监测反应完全后,将体系减压过滤,滤液经真空浓缩、以纯石油醚、200-300硅胶柱层析得1.72g中间体ⅰ,产率62.68%。
[0052]
中间体ⅰ的物性和核磁结果如下:
[0053]
3-溴-5-(3,5-二氯苯基)-5-(三氟甲基)-4,5-二氢异恶唑的合成,白色固体。
[0054]1hnmr(400mhz,dmso-d6)δ7.80(t,j=1.9hz,1h,arh),7.57(d,j=1.8hz,2h,arh),4.24(d,j=18.7hz,1h,ch),4.17(d,j=18.8hz,1h,ch).
[0055]
2、中间体ⅱ的制备:
[0056][0057]
将中间体ⅰ(100mg,0.28mmol)、化合物c(83mg,0.32mmol)、无水碳酸钾(77mg,0.56mmol)和2ml无水四氢呋喃加入50ml双口瓶。以氩气置换瓶内空气三次后,加入四(三苯基磷)钯(6mg,0.0052mmol),在60℃下搅拌反应过夜。tlc(石油醚:乙酸乙酯:乙酸=4:1:1滴)监测反应完全后,将混合物倒入15ml水中,以乙酸乙酯(10ml
×
3)萃取,再用10ml饱和氯化钠溶液洗涤,收集的有机相经无水硫酸钠干燥、真空浓缩、柱层析得105mg中间体ⅱ,产率88.88%。
[0058]
中间体ⅱ的物性和核磁结果如下:
[0059]
4-(5-(3,5-二氯苯基)-5-(三氟甲基)-4,5-二氢异恶唑-3-基)-2-甲基苯甲酸甲酯,白色固体。
[0060]1hnmr(400mhz,dmso-d6)δ13.08(s,1h,cooh),7.89
–
7.86(m,1h,arh),7.78(t,j=1.9hz,1h,arh),7.65
–
7.60(m,4h,arh),4.38(d,j=18.4hz,1h,ch),4.29(d,j=18.4hz,1h,ch),2.54(s,3h,ch3);ms(esi):m/z418.95(m+h)
+
.
[0061]
3、弗雷拉纳的合成:
[0062][0063]
将中间体ⅱ(105mg,0.25mmol)、化合物b(52mg,0.27mmol)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(58mg,0.30mmol)、4-二甲氨基吡啶(62mg,0.50mmol)和2ml二氯甲烷加入25ml烧瓶,在室温下搅拌反应过夜。tlc(石油醚:乙酸乙酯=2:1)监测反应完全后,将体系柱层析得124mg的弗雷拉纳,产率87.22%。
[0064]
弗雷拉纳的物性和核磁结果如下:
[0065]
白色固体。
[0066]1hnmr(400mhz,cdcl3)δ7.53
–
7.46(m,6h,arh),7.26(d,j=2.8hz,1h,nh),7.03(d,j=3.9hz,1h,nh),4.20(dd,j=5.1,2.7hz,2h,ch2),4.08(d,j=17.3hz,1h,ch),3.95
–
3.86(m,2h,ch2),3.70(d,j=17.2hz,1h,ch),2.44(s,3h,ch3);ms(esi):m/z556.10(m+h)
+
.
[0067]
对比例
[0068]
本对比例的弗雷拉纳的制备路线如下:
[0069][0070]
制备包括如下步骤:
[0071]
1、化合物3的合成:
[0072]
在100ml的圆底烧瓶中依次加入2.00g(0.0088mol)的原料1、2.33g(0.0097mol)的原料2和30ml的dmf,搅拌反应至反应液变澄清。用氩气置换三次,加入1.20g(0.012mol)的三乙胺,反应液立即变浑浊,60℃下反应8h。tlc检测反应完毕后,加入20ml蒸馏水继续搅拌1h,用乙酸乙酯萃取三次,每次30ml,合并萃取有机相分别用蒸馏水和饱和食盐水洗涤三次,每次20ml,用无水硫酸钠干燥,过滤,浓缩滤液。经硅胶柱层析(洗脱剂为石油醚:乙酸乙酯=25:1)分离得到3.01g的化合物3.产率79.5%。
[0073]
化合物3的物性和核磁结果如下:
[0074]
白色固体。
[0075]1hnmr(400mhz,cdcl3)δ7.95(d,j=8.6hz,1h,arh),7.56-7.46(m,4h,arh),7.42(t,j=1.8hz,1h,arh),4.10(d,j=17.3hz,1h,ch),3.91(s,3h,och3),3.71(d,j=17.3hz,1h,ch),2.62(s,3h,ch3);ms(esi),2.44(s,3h,ch3);ms(esi):m/z318.0(m+h)
+
.
[0076]
2、化合物4的合成
[0077]
称取1.00g(0.0023mol)化合物3于100ml单口烧瓶中,加入15ml甲醇溶解。取20ml的4n氢氧化钠溶液,缓慢滴加到上述溶液中,并将反应液在60℃下反应4h。tcl检测反应完全后,用乙酸乙酯萃取三次,每次30ml,合并有机相,用饱和食盐水洗涤三次,每次20ml,无水硫酸钠干燥后过滤,浓缩,得到0.86g的化合物4,产率88.9%。
[0078]
化合物4的物性和核磁结果如下:
[0079]
白色固体。
[0080]1hnmr(400mhz,dmso-d6)δ13.08(s,1h,cooh),7.89-7.86(m,1h,arh),7.78(t,j=1.9hz,1h,arh),7.65-7.60(m,4h,arh),4.38(d,j=18.4hz,1h,ch),4.29(d,j=18.4hz,1h,ch),2.54(s,3h,ch3);ms(esi),2.44(s,3h,ch3);ms(esi):m/z418.95(m+h)
+
.
[0081]
3、弗雷拉纳的合成:
[0082]
称取0.05g(0.0012mol)化合物4于100ml单口烧瓶中,用10ml的dmf溶解,加入0.33g(0.0012mol)的dppa,冰浴条件下搅拌,缓慢滴加0.24g(0.0024mol)三乙胺,冰浴下反应1h。将反应液移至室温下反应,分批加入0.19g(0.0012mol)2-氨基-n-(2,2,2-三氟乙
基)-乙酰胺盐酸盐,继续反应12h。tlc检测反应完全后,加入20ml蒸馏水继续搅拌20min,用乙酸乙酯萃取三次,每次30ml,合并有机相分别用蒸馏水和饱和食盐水洗涤三次,每次20ml,用无水硫酸钠干燥,过滤并浓缩滤液,得到灰色粗产物,粗产物用石油醚/乙酸乙酯混合溶剂重结晶,得到0.56g的弗雷拉纳,产率85.2%。
[0083]
弗雷拉纳的物性和核磁结果如下:
[0084]
白色固体。
[0085]1hnmr(400mhz,cdcl3)δ7.53
–
7.46(m,6h,arh),7.26(d,j=2.8hz,1h,nh),7.03(d,j=3.9hz,1h,nh),4.20(dd,j=5.1,2.7hz,2h,ch2),4.08(d,j=17.3hz,1h,ch),3.95
–
3.86(m,2h,ch2),3.70(d,j=17.2hz,1h,ch),2.44(s,3h,ch3);ms(esi):m/z556.10(m+h)
+
.
[0086]
实施例与对比例相比:实施例提升了弗雷拉纳的产率,对比例制备的弗雷拉纳的产率为85.2%,小于实施例的产率87.22%。
[0087]
本实施例中采用1,3-二氯-5-(1-三氟甲基-乙烯基)-苯和二溴甲醛肟能够优先合成得到含有异恶唑环的中间体,通过suzuki-miyaura反应和酰胺化缩合反应能够制得弗雷拉纳,各原料的成本低,来源途径简单。
[0088]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1