一种1-氯-2,3,3-三氟丙烯的制备方法与流程
本发明涉及氟氯烯烃的制备,特别涉及一种以2-氯-1,1-二氟乙烷(hcfc-142)为原料,依次经裂解、调聚脱氯和液相氟化三步反应制备获得1-氯-2,3,3-三氟丙烯的方法。
背景技术:
1、2-氯-1,1-二氟乙烷(hcfc-142)为工业上二氟乙烷(hfc-152a)光氯化制备1-氯-1,1-二氟乙烷(hcfc-142b)的副产,目前在产业上多为燃烧处理,也有研究提出对hcfc-142进行资源化利用,如专利cn109180420a中提出,可将hcfc-142副产转化为偏氟乙烯,但hcfc-142的转化率最高仅51%,hcfc-142的利用率还有待提升。
2、1-氯-2,3,3-三氟丙烯(简称hcfo-1233yd),具有顺反同分异构体hcfo-1233yd(z)(沸点54℃)和hcfo-1233yd(e)(沸点48℃),其臭氧消耗潜值(odp)接近于0,大气寿命约为2.3天,gwp100=0.016,无闪点,表面张力和粘度也很低,与矿物油相容性好,具有优良的理化性能,在清洗剂、制冷剂、发泡剂、气溶胶、绝缘材料和阻燃剂等应用领域具有较大的应用前景。
3、目前,hcfo-1233yd的合成研究较少,主要包括以下路线:
4、一、3-氯-1,1,2,2-四氟丙烷(hcfc-244ca)脱氟化氢路线
5、大金专利cn107250088a和agc专利cn201680044085公开了以四氟丙醇为原料,经氯化反应生成hcfc-244ca,再脱氟化氢制备获得hcfo-1233yd(z/e)的方法。脱氟化氢反应在液相和气相中都可以进行,优选在液相中进行,活性炭为催化剂的气相工艺中原料转化率仅为0.79%。在液相法工艺中,采用氢氧化钾、水和季铵盐形成碱性水溶液,在100℃下反应8小时,hcfc-244ca转化率为90%,1233yd选择性为94.8%,另还存在1-氯-3,3-二氟丙炔1.6%,1233zb 2.1%,244fa0.2%。
6、然而,该工艺的起始原料四氟丙醇易燃且成本较高,且反应中使用的亚硫酰氯等氯化试剂属于易腐蚀有毒化学品,同时,反应中需要使用大量无机碱液作催化剂及洗涤剂,这将产生大量的三废,不利于工业化放大生产。此外,反应最终产物中hcfo-1233yd(z)(沸点54℃)、hcfo-1233yd(e)(沸点48℃)和副产3-氯-1,1,2-三氟丙烯(hcfo-1233yc,沸点42℃左右)、1-氯-1,3,3-三氟丙烯(hcfo-1233zb,沸点47℃左右)等同分异构体沸点相近,相对挥发度接近于1,易形成类共沸物,很难通过常规精馏分离提纯,导致生产成本增加。
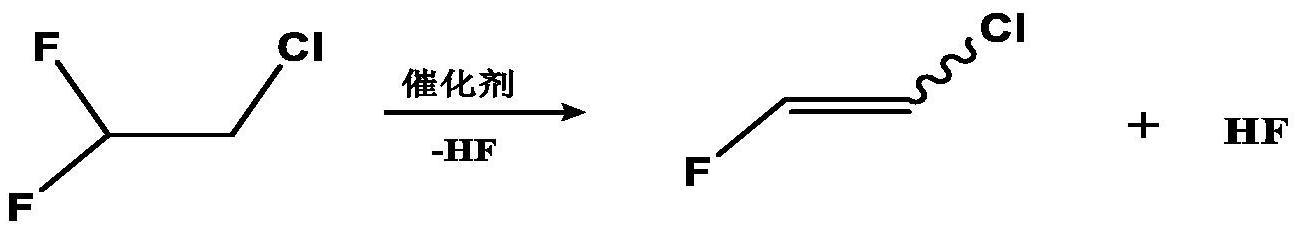
7、二、1,2-二氯-3,3-二氟丙烯(hcfo-1232aa)氟化路线
8、淄博雷玛国际贸易有限公司专利cn112125776a公开了以1,1,2,3,3-五氯丙烷(hcc-240da)为起始原料,经两步氟化获得hcfo-1233yd的方法,具体包括:(1)在铬基催化剂作用下,hcc-240da经氟化氢氟化获得hcfo-1232aa;(2)在氟化氢处理的铬基催化剂为主催化剂,以及zn、co、ni、cu中至少一种为辅催化剂的共同催化作用下,再次氟化获得hcfo-1233yd。步骤(1)反应温度为275℃、空速240h-1、hf:hcc-240da=15:1,hcc-240da转化率为99.6%,hcfo-1232aa选择性为90.4%;步骤(2)反应温度350℃、空速220h-1、hf:hcc-240da=4:1时,hcfo-1232aa转化率为65.16%,hcfo-1233yd的选择性为70.58%。
9、然而,反应起始原料1,1,2,3,3-五氯丙烷(hcc-240da)属于国家重点受控淘汰物质,无法通过采购获得,必须自行制备(如三氯甲烷和二氯乙烯调聚获得),导致反应路线过长。且,两步氟化步骤均采用氟化氢为氟化试剂,而氟化氢不仅具有极强腐蚀性,且常温下为气体,反应工艺中需要精确控制hf/hcc-240da投料比才能获得高选择性的中间体hcfo-1232aa,产业上操作难度较大。
10、三、1,2-二氯-2,3,3-三氟丙烷(hcfc-243ba)脱氯化氢路线
11、agc专利wo2019189024a公开了以hcfc-243ba为原料,在碱液及相转移催化剂存在下,脱氯化氢获得hcfo-1233yd的方法。所述方法具体包括以下步骤:(1)在调聚催化剂作用下,二氯乙烯和三氯甲烷调聚产物240da;(2)240da在碱液作用下脱氯化氢得到1230xd;(3)1230xd在气相氟氯交换反应催化剂作用下,氟氯交换得到1232xd;(4)1232xd在氟化催化剂作用下,氟化制备243ba;(5)243ba在碱液作用下,脱氯化氢得到hcfo-1233yd。具体地,步骤(1)在alcl3作用下,40-45℃调聚反应28h得到240da,240da收率为87.6%;步骤(2)48wt%naoh碱催化剂、19.8gtbab(四丁基溴化铵,相移剂)、50℃、反应2.5h收得纯度99.9%1230xd产物;步骤(3)sbf3氟氯交换催化剂、130℃气相氟氯交换反应得到收率为53%的1232xd产物;步骤(4)sbcl5氟化催化剂、80-90℃、0.95mpa、反应5h得到收率为35.6%的243ba产物;步骤(5)40%的koh碱催化剂、1.01gtbac(四丁基氯化铵,相移催化剂)、50℃液相法脱氯化氢,243ba转化率为98.2%、hcfo-1233yd(z)和hcfo-1233yd(e)选择性分别为94.2%、5.7%。然而,该方法采用了5步法得到hcfo-1233yd(z),工艺路线长、反应条件较难控制,通过反应步骤(1)-(4)中四步法制备中间体243ba的实际收率只有35%左右。
12、因此,从工业化角度分析,上述三条工艺路线或原料不易得,或操作难度较大,不具有放大生产的操作性和经济性。
技术实现思路
1、为了解决上述技术问题,本发明提出了一种原料易得、成本低,产物选择性高、纯度高且易分离,尤其适用于产业化生产的1-氯-2,3,3-三氟丙烯的制备方法。
2、本发明的目的是通过以下技术方案实现的:
3、一种1-氯-2,3,3-三氟丙烯的制备方法,所述制备方法包括:
4、(1)裂解步骤:2-氯-1,1-二氟乙烷(hcfc-142)在裂解催化剂作用下,经裂解脱氟化氢获得1-氯-2-氟乙烯(hcfo-1131),反应式如下:
5、
6、(2)调聚脱氯步骤:在催化剂和促进剂的共同作用下,1-氯-2-氟乙烯和二氯氟甲烷经一步调聚脱氯反应获得1,2-二氯-3,3-二氟丙烯反应式如下:
7、
8、所述催化剂为lewis酸碱双组分催化剂,第一组分选自al、sn、zr、hf、nb、ti、ga或cu中至少一种的金属氯化物,第二组分选自zn、cd、mg、sr、ba、k或cs中至少一种的金属氟化物或金属氯化物;所述促进剂为三氯甲烷和/或二氟氯甲烷;
9、(3)氟化步骤:1,2-二氯-3,3-二氟丙烯在氟化试剂作用下,经液相氟化反应获得1-氯-2,3,3-三氟丙烯(hcfo-1233yd),反应式如下:
10、
11、步骤(1)中,所述裂解催化剂为铝基催化剂,选自铝基氧化物、铝基氟化物或铝基氯化物中的至少一种。优选地,所述裂解催化剂为al-mg复合氧化物和/或氟化物、al-ba复合氧化物和/或氟化物中的至少一种。为了增加反应活性位,提高催化剂的稳定性,所述裂解催化剂可以负载在酸性的活性炭表面。
12、步骤(1)中,所述裂解反应可以是气相反应或液相反应,优选气相催化裂解脱氟化氢反应,裂解温度为300~500℃,原料空速为200~1000h-1,裂解时间为6~10h。原料2-氯-1,1-二氟乙烷经气化室气化后进样,气化室温度为50~80℃;同时加热原料进料管,温度控制为50~80℃。
13、步骤(1)获得的1-氯-2-氟乙烯为顺式-1-氯-2-氟乙烯和/或反式1-氯-2-氟乙烯。
14、步骤(2)中,所述第一组分选自zr和/或nb的金属氯化物,第二组分选自zn、cs或mg中至少一种的金属氟化物或金属氯化物。
15、步骤(2)中,所述促进剂优选为三氯甲烷和二氟氯甲烷。在反应过程中,三氯甲烷在通入反应原料前加入反应器,二氟氯甲烷在通入反应原料后加入反应器。所述反应器优选蒙乃尔400材质的聚合反应釜。
16、步骤(2)中,所述调聚脱氯反应的反应温度为50~150℃,反应压力为0.2~3.0mpa,反应时间为8~24h。优选反应温度为60~120℃,反应压力为0.5~1.5mpa,反应时间为8~12h。更优选反应温度为60~100℃。
17、反应工艺中合适的物料配比,可能获得更优的反应效果。
18、步骤(2)中,所述催化剂、促进剂与二氯氟甲烷的摩尔配比为(0.001~0.5):(0.5~1):1。优选地,催化剂、促进剂与二氯氟甲烷的摩尔配比为(0.01~0.5):(0.8~1):1。
19、步骤(2)中,1-氯-2-氟乙烯和二氯氟甲烷的摩尔配比为1:(0.1~10)。优选地,1-氯-2-氟乙烯和二氯氟甲烷的摩尔配比为1:(1~5)。
20、本发明采用lewis酸碱双组分催化剂和促进剂共同作用,实现了液相调聚和脱卤一步进行,工艺简单、原料易得。本发明在研究过程中发现:调聚反应中活性位为酸性位,而脱氯化氢反应为碱性位,在反应过程中,lewis酸碱双组分催化剂中的酸性组分会先活化反应原料中二氯氟甲烷,使其解离成chfcl+、chcl2+、f-、cl-四种自由基。而二氯氟甲烷在酸性催化剂中活化能垒较低(仅0.79ev),容易发生歧化反应生成chcl3和chf2cl。因此本发明加入三氯甲烷和/或二氟氯甲烷作为促进剂,可以抑制二氯氟甲烷歧化反应的进行,促进二氯氟甲烷解离后的自由基能够选择性加成到氯氟烯烃分子特定位置上,生成饱和的氯氟烷烃分子。而饱和氯氟烷烃遇到碱性活性位会发生脱氯化氢反应,从而生成了目标产物1,2-二氯-3,3-二氟丙烯。
21、将本发明所述调聚脱氯反应的反应液过滤分离,液体部分经精馏或闪蒸可获得1,2-二氯-3,3-二氟丙烯中间体。同时,反应体系中少量未反应的饱和氯氟烷烃产物也可以在闪蒸后进行脱氯得到1,2-二氯-3,3-二氟丙烯。
22、步骤(3)中,所述氟化试剂选自氟化钾(kf)、氟化铯(csf)、氟化钠(naf)、氟化锂(lif)、四丁基氟化铵(tbaf)、氟氢化钾(khf2)中的至少一种。优选地,所述氟化试剂选自kf、csf、tbaf中的至少一种。
23、步骤(3)中,为了增加无机氟化盐在有机物中溶解性,促进金属盐中阳离子极化,氟化反应在极性非质子溶剂中进行,所述极性非质子溶剂选自二甲亚砜(dmso)、环丁砜、n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n-甲基吡咯烷酮(nmp)或乙腈中的至少一种。
24、步骤(3)中,为了促进无机氟化盐中氟离子释放,发生亲核氟化反应,采用相移催化剂作为助剂进行反应,所述相转移催化剂选自18-冠-6-醚、四丁基溴化铵、四丁基氯化铵、季鏻盐中的至少一种。
25、步骤(3)中,1,2-二氯-3,3-二氟丙烯和氟化试剂的摩尔比为1:(1~1.5),反应温度为70~180℃,反应时间为6~24h。
26、本发明以2-氯-1,1-二氟乙烷(hcfc-142)为原料,三步法制备获得hcfo-1233yd,具体包括以下操作步骤:
27、s1.原料hcfc-142经过气化室气化后进入气相裂解反应器中,在裂解反应催化剂床层表面发生脱氟化氢反应生成1-氯-2-氟乙烯(hcfo-1131);收集反应产物,分级精馏,提纯获得1-氯-2-氟乙烯中间体;
28、s2.在lewis酸碱双组分催化剂和促进剂的协同作用下,1-氯-2-氟乙烯与二氯氟甲烷(r21)在聚合反应釜中经一步调聚脱氯化氢获得1,2-二氯-3,3-二氟丙烯;利用气袋收集未反应的原料1-氯-2氟乙烯和二氯氟甲烷,以及低沸促进剂二氟氯甲烷;对反应釜中反应液进行过滤分离,固体部分为lewis酸碱双组分催化剂,液体部分通过精馏或闪蒸得到1,2-二氯-3,3-二氟丙烯;
29、s3.1,2-二氯-3,3-二氟丙烯和氟化试剂在极性非质子溶剂中,在相转移催化剂存在下进行液相氟化反应,反应结束后,反应产物粗品经分液、蒸馏或闪蒸处理后获得目标产品hcfo-1233yd。
30、与现有技术相比,本发明具有的有益效果为:
31、1、本发明的起始原料2-氯-1,1-二氟乙烷(hcfc-142)为工业上二氟乙烷(hfc-152a)光氯化制备二氟一氯乙烷(hcfc-142b)的副产,不仅可以将副产转化为高附加值、环境友好的含氟烯烃单体,还可以大大减少企业生产和副产处理成本。同时,本发明还提高了副产hcfc-142的利用率。
32、2、本发明以hcfc-142为原料,经三步反应获得hcfo-1233yd,相较于现有的2,2,3,3-四氟丙醇两步法工艺,原料成本低、产物选择性高、原料转化率高、产物分离较易,本技术的工艺尤其适用于工业化生产。
33、3、本发明第三步液相氟化工艺,氟化试剂廉价易得,反应温度低,反应条件易于控制,且反应产物沸点相差大,通过普通精馏提纯工艺,即可获得纯度≥99.5%的高纯产品,并可实现未反应原料的回收再利用。
- 还没有人留言评论。精彩留言会获得点赞!