一种N-苯基-N-三氯甲硫基苯磺酰胺的合成方法与流程
一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法
技术领域
1.本发明涉及有机合成技术领域,尤其涉及一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法。
背景技术:
2.在我们的日常生活中,橡胶材料的应用非常广泛,如我们现在所使用轮胎、胶管等都是由橡胶材料制成。硫化是使橡胶材料具有使用价值所经历的必要过程之一,并且硫化过程直接决定了橡胶制品的使用性能。但是胶料在贮存和加工过程中因受热或其他原因,导致胶料在发生正常的硫化反应之前就已经发生了部分交联,失去原本的加工流动性和再加工能力,这种现象就是我们常说的焦烧现象。烧焦问题可以通过调整工艺条件、调整胶化体系来解决,但是在实际生产过程中,工艺和胶化体系的改变都会对胶料的性能产生非常大的影响,还需要复杂的设备装置,因此,选择合适的防焦剂是防止胶料焦烧的最为简单可行的方法。
3.目前,常用的防焦剂主要有三种,分别是有机酸类、亚硝基化合物和次磺酰胺类。有机酸类和亚硝基化合物的防焦剂存在防焦能力弱、对促进剂和胶种的选择性大,易出现分散不良、延迟硫化等问题。次磺酰胺类防焦剂是含有s-n键合的一系列化合物,其中n-苯基-n-三氯甲硫基苯磺酰胺(防焦剂e)能避免上述问题,在同样的配合体系下,对胶料的硫化特性与物理性能无不良影响,贮存稳定性高、操作性良好,符合工业生产安全和卫生要求,是一种理想的防焦剂。
4.目前主要采用两步法制备防焦剂e,在制备过程中,其中间体n-苯基苯磺酰胺必须在强碱性条件进行制备,且反应温度必须控制在-15℃以下,反应条件苛刻,导致反应能耗较高;此外,该中间体n-苯基苯磺酰胺常采用120#溶剂油等有机溶剂进行萃取,不能彻底消除进入溶剂油体系的苯胺或苯胺盐酸盐,导致产品纯度较低,需要额外的后处理工艺进行提纯。溶剂油等有机溶剂虽然可以通过蒸馏回收后再利用,但由全氯甲硫醇产生的二硫化碳和四氯化碳等杂质在溶剂油中不断累积,导致最终产生的废液难以回收处理,造成环境污染,而且废液中含有的高度易燃的低沸点组分更是存在较大的安全隐患。有研究者认为采用甲苯为溶剂,可解决反应条件苛刻的问题,但是其制备的n-苯基苯磺酰胺中含有残余的甲苯或其衍生物,导致其纯度难以满足防焦剂e的制备要求。有研究者认为加入四丁基溴化铵作为催化剂可缩短反应时间,但是在反应过程中需要反复升温降温,导致能耗较高,中间体的含量和收率均偏低。
技术实现要素:
5.针对现有的n-苯基-n-三氯甲硫基苯磺酰胺反应条件苛刻、能耗高、收率较低、纯度低、存在安全隐患等问题,本发明提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法。
6.为达到上述发明目的,本发明实施例采用了如下的技术方案:
7.一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,所述合成方法包括以下步骤:
8.步骤一、将苯胺、4-二甲氨基吡啶和水混合均匀,得乳化液;
9.步骤二、在10℃~25℃条件下,将苯磺酰氯滴加至所述乳化液中,滴加完毕后,保温反应10h~20h,当反应体系中ph为6~7时,反应结束,过滤,得中间体;
10.步骤三、于-5℃~0℃条件下将所述中间体、4-二甲氨基吡啶和液碱混合均匀,再于-5℃~5℃条件下,滴加全氯甲硫醇,滴加完毕后,再于20℃~100℃条件下水解反应1h~2h,离心,收集滤饼,清洗,烘干,得到n-苯基-n-三氯甲硫基苯磺酰胺。
11.上述合成方法中的反应过程如下:
[0012][0013]
相对于现有技术,本技术提供的n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,具有以下优势:
[0014]
本技术以4-二甲氨基吡啶为催化剂,促进苯胺与苯磺酰氯之间反应的进行,并促进中间体与全氯甲硫醇反应的进行,缩短反应时间,提高反应收率;采用水相的反应体系,能彻底解决残留苯胺及其形成的苯胺盐酸盐水溶液进入下一步体系中引发副反应的问题,还能避免溶剂燃烧爆炸等安全隐患以及有机溶剂尾气造成的污染等问题,因此本技术采用温和的反应条件制备得到高纯度的中间体和产物,降低能耗,省去安全环保的配套装置,显著降低生产成本。
[0015]
上述步骤二中洗涤滤饼的水,可用于回收套用,能代替步骤一中的水,用于制备乳化液;抽滤的滤液可用于制备副产苯胺盐酸盐,也可以加碱后蒸馏回收苯胺。上述步骤三中洗涤滤饼的水,也能用于回收套用,可用于步骤三中液碱的制备。本技术提供的合成方法中产生的废水,能用于回收套用,达到了清洁生产的效果。
[0016]
本技术采用特定的催化剂4-二甲氨基吡啶和水相的反应体系,反应条件温和,过程容易控制,反应时间短,收率高,纯度高,且废水还能进行回收套用,安全环保,有利于进行工业化推广和应用。
[0017]
可选的,步骤一中,所述苯胺、4-二甲氨基吡啶和水的质量比为45:1.5~2.5:250~300。
[0018]
可选的,所述苯胺与所述苯磺酰氯的质量比为8.8~9.2:7.8~8.2。
[0019]
优选的各组分比例,使得反应充分进行,且微过量的苯胺还能作为缚酸剂,且不破坏体系的酸性环境,避免苯磺酰氯的水解,提高反应收率和纯度。
[0020]
可选的,所述苯磺酰氯的滴加时间为2h~3h。
[0021]
优选的苯磺酰氯的滴加速度使得反应原料充分进行,并避免苯磺酰氯的水解,提高反应收率。
[0022]
可选的,所述液碱的浓度为3wt%~3.5wt%。
[0023]
可选的,步骤三中,所述苯磺酰氯、4-二甲氨基吡啶和液碱的质量比为1:0.03~0.05:11~12.5。
[0024]
可选的,所述全氯甲硫醇与所述苯磺酰氯的质量比为1.28~1.32:1。
[0025]
优选的各组分之间的比例,使得反应充分进行,避免副产物的产生,提高产品的纯度。
[0026]
可选的,所述水解反应的温度为40℃~70℃。
[0027]
可选的,所述全氯甲硫醇的滴加时间为3h~4h。
[0028]
优选的全氯甲硫醇的滴加速度使得反应原料充分进行,并避免体系酸性过大,阻碍反应的进行。
[0029]
可选的,所述n-苯基-n-三氯甲硫基苯磺酰胺还包括粉粹,且所述粉碎的细度为120目~300目。
[0030]
防焦剂n-苯基-n-三氯甲硫基苯磺酰胺还可粉碎至不同细度,以适用于不同胶料体系。
附图说明
[0031]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0032]
图1是本发明实施例1提供的dsc测试图;
[0033]
图2是本发明实施例1提供的红外光谱图;
[0034]
图3是本发明实施例2提供的dsc测试图;
[0035]
图4是本发明实施例2提供的红外光谱图;
[0036]
图5是本发明实施例3提供的dsc测试图;
[0037]
图6是本发明实施例3提供的红外光谱图;
[0038]
图7是本发明实施例4提供的dsc测试图;
[0039]
图8是本发明实施例4提供的红外光谱图;
[0040]
图9是本发明对比例1提供的红外光谱图。
具体实施方式
[0041]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0042]
实施例1
[0043]
本发明实施例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,所述合成方法包括以下步骤:
[0044]
步骤一、将苯胺45g、4-二甲氨基吡啶2g和水300ml混合均匀,得乳化液;
[0045]
步骤二、在20℃条件下,将苯磺酰氯40g滴加至所述乳化液中,滴加时间为3h,滴加完毕后,保温反应16h,当反应体系中ph为6时,反应结束,过滤,100ml水冲洗,烘干,得中间体n-苯基苯磺酰胺52.5g,含量99.6%,收率99.0%;
[0046]
步骤三、于-3℃条件下将所述中间体n-苯基苯磺酰胺52.5g、4-二甲氨基吡啶1.5g
和浓度为3wt%的液碱470g全部溶解混合均匀,再于0℃条件下,滴加全氯甲硫醇52.2g,滴加时间为3.5h,滴加完毕后,再升温至20℃,检测体系的ph值为8,水解反应1h,至体系的ph值为6,再升温至80℃水解反应1h,抽滤,收集滤饼,清洗,烘干,得到n-苯基-n-三氯甲硫基苯磺酰胺82.3g,含量为99.7%,收率为94.7%。
[0047]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺进行dsc检测,检测结果如图1所示,从图1中可以看出n-苯基-n-三氯甲硫基苯磺酰胺的熔程111.75℃~117.11℃,峰值温度为113.96℃,由此可知,本技术制备的n-苯基-n-三氯甲硫基苯磺酰胺熔点高,熔程短。
[0048]
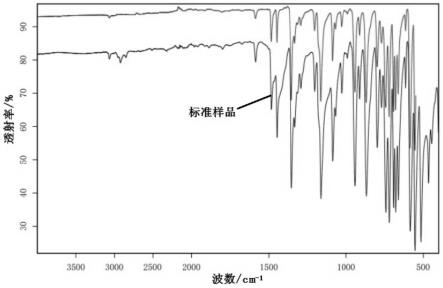
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺与其标准样品进行红外分析,结果如图2所示,从图2中可以看出,实施例1制备的n-苯基-n-三氯甲硫基苯磺酰胺与标准样品的红外光谱匹配度为907,由此可知,本技术制备的产品即为n-苯基-n-三氯甲硫基苯磺酰胺。
[0049]
实施例2
[0050]
本发明实施例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,所述合成方法包括以下步骤:
[0051]
步骤一、将苯胺47g、4-二甲氨基吡啶2.5g和水280ml混合均匀,得乳化液,其中上述水包括之前中间体n-苯基苯磺酰胺冲洗的废液;
[0052]
步骤二、在15℃条件下,将苯磺酰氯40g滴加至所述乳化液中,滴加时间为2.5h,滴加完毕后,保温反应14h,当反应体系中ph为7时,反应结束,过滤,冲洗,烘干,得中间体n-苯基苯磺酰胺52.4g,含量99.5%,收率98.7%;
[0053]
步骤三、于-5℃条件下将所述中间体n-苯基苯磺酰胺52.4g、4-二甲氨基吡啶1.26g和浓度为3.5wt%的液碱462g混合均匀,再于-5℃条件下,滴加全氯甲硫醇53.8g,滴加时间为3.5h,滴加完毕后,再升温至20℃,检测体系的ph值为8,水解反应1h,至体系的ph值为6,再升温至60℃水解反应1h,抽滤,收集滤饼,清洗,烘干,得到n-苯基-n-三氯甲硫基苯磺酰胺82.1g,含量为99.4%,收率为94.2%。
[0054]
上述3.5wt%的液碱采用n-苯基-n-三氯甲硫基苯磺酰胺冲洗废液配制得到。
[0055]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺进行dsc检测,检测结果如图3所示,从图3中可以看出n-苯基-n-三氯甲硫基苯磺酰胺的熔程110.75℃~114.94℃,峰值温度为112.76℃,由此可知,本技术制备的n-苯基-n-三氯甲硫基苯磺酰胺熔点高,熔程短。
[0056]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺与其标准样品进行红外分析,结果如图4所示,从图4中可以看出,实施例2制备的n-苯基-n-三氯甲硫基苯磺酰胺与标准样品的红外光谱匹配度为865,由此可知,本技术制备的产品即为n-苯基-n-三氯甲硫基苯磺酰胺。
[0057]
实施例3
[0058]
本发明实施例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,所述合成方法包括以下步骤:
[0059]
步骤一、将苯胺43g、4-二甲氨基吡啶1.5g和水250ml混合均匀,得乳化液;
[0060]
步骤二、在10℃条件下,将苯磺酰氯40g滴加至所述乳化液中,滴加时间为2h,滴加完毕后,保温反应20h,当反应体系中ph为6.5时,反应结束,过滤,得中间体n-苯基苯磺酰胺湿料;
[0061]
步骤三、于0℃条件下将所述n-苯基苯磺酰胺湿料、4-二甲氨基吡啶1.6g和浓度为3wt%的液碱500g混合均匀,再于5℃条件下,滴加全氯甲硫醇52.8g,滴加时间为4h,滴加完毕后,再升温至20℃,检测体系的ph值为8,水解反应1h,至体系的ph值为6,再升温至40℃水解反应1h,抽滤,收集滤饼,清洗,烘干,得到n-苯基-n-三氯甲硫基苯磺酰胺81.6g,含量为99.3%,收率为93.6%。
[0062]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺进行dsc检测,检测结果如图5所示,从图5中可以看出n-苯基-n-三氯甲硫基苯磺酰胺的熔程111.11℃~116.67℃,峰值温度为113.71℃,由此可知,本技术制备的n-苯基-n-三氯甲硫基苯磺酰胺熔点高,熔程短。
[0063]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺与其标准样品进行红外分析,结果如图6所示,从图6中可以看出,实施例3制备的n-苯基-n-三氯甲硫基苯磺酰胺与标准样品的红外光谱匹配度为905,由此可知,本技术制备的产品即为n-苯基-n-三氯甲硫基苯磺酰胺。
[0064]
实施例4
[0065]
本发明实施例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,所述合成方法包括以下步骤:
[0066]
步骤一、将苯胺45g、4-二甲氨基吡啶2.2g和水300ml混合均匀,得乳化液;
[0067]
步骤二、在25℃条件下,将苯磺酰氯40g滴加至所述乳化液中,滴加时间为3h,滴加完毕后,保温反应15h,当反应体系中ph为6时,反应结束,过滤,清洗,得中间体n-苯基苯磺酰胺湿料;
[0068]
步骤三、于0℃条件下将所述中间体n-苯基苯磺酰胺湿料、4-二甲氨基吡啶1.2g和浓度为3wt%的液碱460g混合均匀,再于0℃条件下,滴加全氯甲硫醇52g,滴加时间为3h,滴加完毕后,再升温至20℃,水解反应2h,离心,收集滤饼,清洗,烘干,得到n-苯基-n-三氯甲硫基苯磺酰胺82.6g,含量为99.1%,收率为94.5%。
[0069]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺进行dsc检测,检测结果如图7所示,从图7中可以看出n-苯基-n-三氯甲硫基苯磺酰胺的熔程110.13℃~115.21℃,峰值温度为112.51℃,由此可知,本技术制备的n-苯基-n-三氯甲硫基苯磺酰胺熔点高,熔程短。
[0070]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺与其标准样品进行红外分析,结果如图8所示,从图8中可以看出,实施例4制备的n-苯基-n-三氯甲硫基苯磺酰胺与标准样品的红外光谱匹配度为902,由此可知,本技术制备的产品即为n-苯基-n-三氯甲硫基苯磺酰胺。
[0071]
为了更好的说明本发明的技术方案,下面还通过对比例和本发明的实施例做进一步的对比。
[0072]
对比例1
[0073]
本对比例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,将步骤一种的水替换为甲苯,其余条件与实施例1一致,不再赘述。
[0074]
本对比例提供的合成方法中,中间体n-苯基苯磺酰胺为38.8g,含量97.5%,收率71.6%;产品n-苯基-n-三氯甲硫基苯磺酰胺59.7g,含量为94.2%,收率为64.9%。
[0075]
将上述制备的n-苯基-n-三氯甲硫基苯磺酰胺与其标准样品进行红外分析,结果如图9所示,从图9中可以看出,对比例1制备的n-苯基-n-三氯甲硫基苯磺酰胺与标准样品
的红外光谱匹配度为677,且在3000cm-1
~3100cm-1
处出现了c-h的振动峰,该振动峰是由甲苯及其衍生物中的ch
3-造成的,由此可知,若是采用有机溶剂体系,容易引进杂质,进而降低产品的纯度和收率。
[0076]
对比例2
[0077]
本对比例提供一种n-苯基-n-三氯甲硫基苯磺酰胺的合成方法,将步骤一和步骤三中的4-二甲氨基吡啶均替换为三乙基苄基氯化胺,其余条件与实施例1一致,不再赘述。
[0078]
本对比例提供的合成方法中,中间体n-苯基苯磺酰胺为47.7g,含量98.1%,收率88.6%;产品n-苯基-n-三氯甲硫基苯磺酰胺73.5g,含量为95.5%,收率为81.0%。
[0079]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1