二咔唑苯基双膦配体及其制备方法、咔唑苯基双膦卤化亚铜及其制备方法和应用
1.本发明属于铜配合物技术领域,具体涉及一种4,5-二咔唑苯基双膦配体及其制备方法、咔唑苯基双膦卤化亚铜及其制备方法和应用。
背景技术:
2.亚铜配合物相比贵金属铱、铂配合物具有成本低、环境友好、良好的发光性质,具有替代贵金属磷光配合物的潜力。尤其是亚铜配合物中低能级mlct激发态具有较小的单、三重态能隙,能有效俘获三重态激子用于热激活延迟荧光(tadf),应用于高效有机发光二极管(oleds)。
3.近年来,含有机双膦的卤化亚铜配合物因其结构的刚性,可降低激发态构型发生jahn-teller形变导致的能量损耗,可获得高量子效率的亚铜配合物。咔唑是一个具有刚性结构、大的能级差的高效蓝光材料,具有良好的空穴传输能力,有必要对该技术方案进一步的进行探索和研发。
技术实现要素:
4.为解决现有技术的不足,本发明提供了一种4,5-二咔唑苯基双膦配体及其制备方法、咔唑苯基双膦卤化亚铜及其制备方法和应用。本发明所提供的咔唑苯基双膦卤化亚铜发光寿命小于3微秒、高效。
5.本发明所提供的技术方案如下:
6.一种4,5-二咔唑苯基双膦配体,结构式如下:
[0007][0008]
本发明所提供的1,2-二咔唑基-4,5-二(二苯基膦基)苯的双齿有机膦配体(dcdp),可与卤化亚铜反应后获得了三个双核四配位卤化亚铜配合物。
[0009]
本发明还提供了上述4,5-二咔唑苯基双膦配体的制备方法,其合成路线如下:
[0010][0011]
本发明还提供了一种咔唑苯基双膦卤化亚铜,结构式如下:
[0012][0013]
本发明所提供的1,2-二咔唑基-4,5-二(二苯基膦基)苯的双齿有机膦配体(dcdp),与卤化亚铜反应后获得了三个双核四配位卤化亚铜配合物,[cu2x2(dcdp)2][dcdp=1,2-二咔唑基-4,5-二(二苯基膦基)苯,x=i(1),br(2),cl(3)]。结构分析表明,2个卤原子与2个铜原子桥连,形成双核四配位卤化亚铜配合物。
[0014]
本发明还提供了上述咔唑苯基双膦卤化亚铜的制备方法,其合成路线如下:
[0015][0016]
整体的合成路线如下:
[0017][0018]
本发明还提供了上述咔唑苯基双膦卤化亚铜的应用,作为荧光材料。
[0019]
进一步的,作为热激活延迟荧光材料。
[0020]
具体的,x为i,所述的咔唑苯基双膦卤化亚铜作为绿黄光荧光材料。
[0021]
具体的,x为br,所述的咔唑苯基双膦卤化亚铜作为黄光荧光材料。
[0022]
具体的,x为cl,所述的咔唑苯基双膦卤化亚铜作为黄橙光荧光材料。
[0023]
本发明所提供的上述配合物1,2和3在室温固态下分别发绿黄光、黄光和黄橙光,最大发射波长分别为567,580和602nm。室温下固态绝对内量子效率φ
pl
为0.09~0.53,发光寿命为1.2-2.3μs。配合物1-3的发光为热激活延迟荧光,主要来自xlct(卤素到配体的电荷跃迁)、mlct(金属到配体的电荷跃迁)和ilct配体内电荷跃迁(配体内电荷跃迁)。
附图说明
[0024]
图1是本发明所提供的配体dcdp在cdcl3中的1h nmr谱。
[0025]
图2是本发明所提供的配合物1在cdcl3中的1h nmr谱。
[0026]
图3是本发明所提供的配合物2在cdcl3中的1h nmr谱。
[0027]
图4是本发明所提供的配合物3在cdcl3中的1h nmr谱。
[0028]
图5是本发明所提供的配体dcdp在cdcl3的
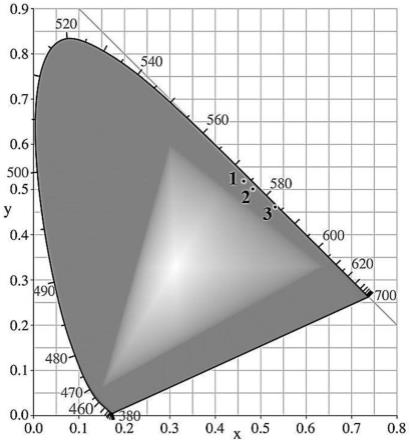
13
c nmr谱。
[0029]
图6是本发明所提供的配体dcdp在cdcl3的
31
p nmr谱。
[0030]
图7是本发明所提供的配合物1在cdcl3的
31
p nmr谱。
[0031]
图8是本发明所提供的配合物2在cdcl3的
31
p nmr谱。
[0032]
图9是本发明所提供的配合物3在cdcl3的
31
p nmr谱。
[0033]
图10是本发明所提供的配体dcdp的质谱。
[0034]
图11是本发明所提供的配合物1的质谱。
[0035]
图12是本发明所提供的配合物2的质谱。
[0036]
图13是本发明所提供的配合物3的质谱。
[0037]
图14是本发明所提供的配合物1-3的ortep图。
[0038]
图15是本发明所提供的298k下配合物1-3和配体dcdp在ch2cl2中的紫外吸收光谱。
[0039]
图16是本发明所提供的tddft计算的配合物1在ch2cl2中的吸收光谱。
[0040]
图17是本发明所提供的tddft计算的配合物2在ch2cl2中的吸收光谱。
[0041]
图18是本发明所提供的tddft计算的配合物3在ch2cl2中的吸收光谱。
[0042]
图19是本发明所提供的配合物1在ch2cl2中的前线轨道图。
[0043]
图20是本发明所提供的配合物2在ch2cl2中的前线轨道图。
[0044]
图21是本发明所提供的配合物3在ch2cl2中的前线轨道图。
[0045]
图22是本发明所提供的配合物1-3在固态下的归一化发射光谱(298k和77k)。
[0046]
图23是本发明所提供的配合物1-3的cie图。
[0047]
图24是本发明所提供的配合物1-3优化s0构型的homo和lumo电子云分布图。
[0048]
图25是本发明所提供的配合物1-3优化s1构型的homo和lumo电子云分布图。
[0049]
图26配合物1-3优化的构型(s0、s1和t1态)。
[0050]
图27图23是本发明所提供的配合物1-3的室温发光寿命(a:1;b:2;c:3)。
具体实施方式
[0051]
以下对本发明的原理和特征进行描述,所举实施例只用于解释本发明,并非用于限定本发明的范围。
[0052]
1.1仪器与试剂
[0053]
试剂:所有试剂均为市售,分析纯。乙醚(et2o)溶剂经钠丝浸泡除水24h后使用。四氢呋喃(thf)经钠丝除水24h后重蒸,并用二苯甲酮作指示剂。
[0054]
仪器:红外光谱采用美国perkin elmet公司bx ft-ir型傅里叶转换红外光谱仪(溴化钾压片),1h,
13
c和
31
p nmr谱用varian 400mhz nmr光谱仪,使用氘带试剂锁场及参比,化学位移以ppm计量,h谱以sime4为标准,磷谱以85%h3po4为标准。高分辨质谱采用hrms-esi质谱仪。配合物的单晶结构采用bruker apex duo衍射仪。紫外可见光谱采用unicam heλiosα光谱仪,光致发光光谱采用fls980稳态和时间分辨荧光光谱仪。固态量子效率采用hamamatsu体系装有积分球测定绝对量子效率。热失重分析采用perkin-elmer diamond tg/dta热分析仪。
[0055]
1.2合成
[0056]
1.2.1配体dcdp的合成
[0057]
将装有1,2-二咔唑基-4,5-二溴苯(2.8315g,5.0mmol)的反应瓶抽真空,通氮气,加入30ml无水乙醚(et2o)和30ml四氢呋喃(thf),-95℃下缓慢滴入6ml正丁基锂的正己烷溶液(2.5mol/l,15mmol),保持温度在-85~-95℃,搅拌反应1.5h后加入氯代二苯基膦(3.8ml,20mmol),继续搅拌2h,缓慢升至室温,加入30ml水猝灭反应,加入二氯甲烷萃取反应液,分液后得到有机相,用无水硫酸钠干燥、过滤,除去溶剂得粗品,用柱层析分离得到白色固体1.85g,产率46.4%。1h nmr(400mhz,cdcl3):δ=7.70(d,j=8.0hz,4h),7.54(t,j=8.0hz,2h),7.36~7.28(m,20h),6.97(t,j=16.0hz,4h),6.88(t,j=16.0hz,4h),6.79(d,j=8.0hz,4h).
31
p nmr(160mhz,cdcl3),δ=-14.92(s).
13
c nmr(100mhz,cdcl3):δ=
144.56,144.47,144.38,139.02,136.54,136.51,136.49,135.71,135.68,135.64,133.95,133.85,133.75,133.70,128.93,128.76,128.73,128.69,125.31,123.48,120.08,119.75,109.52.hrms(esi):m/z calcd for[m+1]
+
,777.2544,found:777.2555.
[0058]
1.2.2配合物1的合成
[0059]
在室温下将碘化亚铜(95.23mg,0.5mmol)加入到溶有dcdp(388.43mg,0.5mmol)的30ml二氯甲烷溶液中,搅拌4小时,过滤,收集滤液并旋干,再用二氯甲烷和正己烷重结晶,真空干燥后得到312.1mg的黄色固体粉末,产率64.5%。1h nmr(400mhz,cdcl3):δ=7.71~7.68(m,12h),7.44~7.40(m,16h),7.23(t,j=8.0hz,8h),7.14(t,j=8.0hz,16h),6.97(t,j=8.0hz,8h),6.88(t,j=8.0hz,8h),6.81(d,j=8.0hz,8h).
31
p nmr(160mhz,cdcl3),δ=-22.43(s).hrms(esi):m/z calcd for[m-2i-cu]
+
,1616.4350,found:1616.4356.
[0060]
1.2.3配合物2的合成
[0061]
配合物2的合成与配合物1相似,在室温下将溴化亚铜(71.7mg,0.5mmol)加入到溶有dcdp(388.43mg,0.5mmol)的30ml二氯甲烷溶液中搅拌,过滤,收集滤液并旋干得到285.5mg的黄色固体粉末,再用二氯甲烷和正己烷重结晶,真空干燥后得到产率62.1%。1h nmr(400mhz,cdcl3):δ=7.72~7.66(m,12h),7.48~7.42(m,16h),7.21(t,j=16.0hz,8h),7.14(t,j=8.0hz,16h),6.97(t,j=8.0hz,8h),6.88(t,j=8.0hz,8h),6.80(d,j=8.0hz,8h).
31
p nmr(160mhz,cdcl3),δ=-19.95(s).hrms(esi):m/zcalcd for[m-2br-cu]
+
,1616.4350,found:1616.4367.
[0062]
1.2.4配合物3的合成
[0063]
配合物3的合成与配合物1相似,在室温下将氯化亚铜(49.5mg,0.5mmol)加入到溶有dcdp(388.43mg,0.5mmol)的30ml二氯甲烷溶液中搅拌,过滤,收集滤液并旋干得到261.3mg的黄色固体粉末,再用二氯甲烷和正己烷重结晶,真空干燥后得到产率59.7%。1h nmr(400mhz,cdcl3):δ=7.72~7.66(m,12h),7.50~7.43(m,16h),7.21(t,j=8.0hz,10h),7.15(t,j=8.0hz,14h),6.98(t,j=8.0hz,8h),6.88(t,j=8.0hz,8h),6.79(d,j=8.0hz,8h).
31
p nmr(160mhz,cdcl3),δ=-18.36(s).hrms(esi):m/zcalcd for[m-2cl-cu]
+
,1616.4350,found:1616.4339.
[0064]
2.结果与讨论
[0065]
2.1合成与结构表征
[0066]
配体dcdp和配合物1-3的合成路线见scheme 1。首先,1,2-二氟-4,5-二溴苯和咔唑混合,碳酸铯碱性条件,n,n-二甲基甲酰胺(dmf)为溶剂,160℃下反应,生成1,2-二咔唑-4,5-二溴苯,产率69.2%,然后与正丁基锂以摩尔比1:2.5在氮气氛下,-95℃的无水乙醚和四氢呋喃的混合溶液中反应生成1,2-二锂-4,5-二咔唑基苯,然后继续与氯代二苯基膦反应得到配体dcdp,产率46.4%。配体dcdp和cux以摩尔比1:1在ch2cl2中反应,得到双核卤化亚铜配合物1-3,产率59.7~64.5%,且配合物在空气中稳定,能溶于二氯甲烷、氯仿等有机溶剂。配合物的结构采用核磁共振、高分辨质谱和单晶x-射线衍射等得到确证。配合物1-3的合成路线如图1所示。
[0067]
2.1.1.1h nmr谱
[0068]
图1~图4分别为配体dcdp和配合物1-3在cdcl3氘代试剂中的核磁氢谱,图中化学位移、积分及峰的裂分情况与结构相符。
[0069]
2.1.2.
13
c nmr谱
[0070]
图6为配体dcdp在氘代氯仿中的核磁碳谱。有13种化学环境不同的碳原子,说明配体结构具有对称性,与结构相符。
[0071]
2.1.3.
31
p nmr谱
[0072]
图7~图10分为配体dcdp和配合物1-3在cdcl3中的核磁磷谱。在配体dcdp中有2个p原子,配合物1-3中分别有4个p原子,4个核磁磷谱图都只有一组信号峰,说明它们的结构对称。
[0073]
2.1.4.hrms-esi图谱
[0074]
采用高分辨电喷雾电离质谱仪(hrms-esi)对配体dcdp和配合物1-3做了表征,图10的质谱图显示:m/z为777.2555是配体dcdp的[m+1]
+
峰,与理论计算值777.2544相符;图11-图13的质谱图谱显示:未发现配合物1-3的分子离子峰,可以看到m/z为1616.4356、1616.4367和1616.4339的碎片离子峰,对应于配合物脱去两个卤素原子、一个铜原子的碎片离子峰[m-2x-cu]
+
,与理论计算值1616.4350相符。
[0075]
2.1.5.晶体结构
[0076]
配合物1-3的结构见图14。晶体数据及部分的键长键角数据列于表1和表2。配合物3中有一个溶剂二氯甲烷分子。晶体数据表明,配合物1-3为双核结构,铜中心为弯曲四面体构型,每个铜原子分别与2个p原子和2个卤素相连,2个卤素分别与2个cu原子形成桥连cu2x2结构单元,cu2x2环沿着x
…
x轴弯曲。两个cux2平面三角形之间的二面角为23.53~37.04
°
。配合物1-3中cu
–
x键长随着卤素范德华半径的增大而增长,配合物1中cu
…
cu之间的距离为配合物2和3中cu
…
cu之间的距离分别为和与铜原子的范德华半径之和相比,表明配合物1中铜与铜之间形成共价键,而配合物2和3中2个铜原子间有弱的作用力。
[0077]
表1.配合物1-3的晶体数据
[0078]
[0079][0080]
表2.配合物1-3的部分键长与键角(
°
)
[0081][0082]
2.2.光物理性质和分子轨道计算
[0083]
图15是配合物1-3及其配体dcdp室温下在ch2cl2溶液中的紫外可见吸收光谱。配体及配合物的浓度为2.5
×
10-5
mol/l。配体dcdp在293nm(ε=3.68
×
104m-1
cm-1
)和339nm(ε=2.35
×
104m-1
cm-1
)有最大吸收峰,这是芳香膦化合物的特征吸收,分别对应于π
→
π*和n
→
π*的跃迁,前者是咔唑或苯环内的π电子到反键π*轨道的跃迁,后者是p原子或氮原子上的孤对电子到咔唑环或苯环的反键π*轨道的跃迁。配合物1-3在291~292nm[ε=(4.96~6.32)
×
104m-1
cm-1
]和337nm[ε=(4.59~5.83)
×
104m-1
cm-1
]处出现强的吸收带,在375~445nm处出现一条较弱的吸收尾带。这种较弱的吸收尾带是由于铜到配体、卤素到配体或配体内的电荷跃迁导致的。通过tddft计算,配合物1~3在二氯甲烷中的吸收光谱见图16~图18,计算结果与实验结果一致。根据配合物1-3的激发态性质(表3~表5,图19-21),配合物1-3的最低激发态的主要贡献来源于homo(最高占有分子轨道)到lumo(lumo:最低非占有分子轨
道)。如图19-21所示,优化s0构型及通过dtf计算的配合物1~3的homo和lumo的分子轨道图说明homo上的电子主要分布在铜、卤素和磷原子上,而lumo上的电子主要分布在配体dcdp的苯环和咔唑的氮环上。因此,我们可以推断配合物1~3的最低激发态是由mlct(金属到配体的电荷跃迁)、xlct(卤素到配体的电荷跃迁)及ilct(配体内的电荷跃迁)组成。
[0084]
表3.计算配合物1在ch2cl2中的激发态
[0085][0086]
表4.计算配合物2在ch2cl2中的激发态
[0087][0088][0089]
表5.计算配合物3在ch2cl2中的激发态
[0090][0091]
图22为配合物1-3(自左向右)在293k和77k下固态发射图谱,表3为最大发射波长、298k和77k的寿命、量子效率及通过x-射线分析获得的结构利用tddft计算的数据。配合物1、2和3在室温下分别发绿黄光、黄光和黄橙光,最大发射波长分别为567,580和602nm(激发
波长为365nm),发射光谱宽、无结构化特征,说明发射激发态具有电荷转移特征。室温空气下固态绝对内量子效率φ
pl
为0.09~0.53。1-3的发射最大波长顺序为3》2》1,与卤素的场强顺序相同(i-》br-》cl-)。基于298k的固态荧光光谱,配合物1-3的色度坐标分别为(0.4624,0.5183)、(0.4832,0.5012)和(0.5326,0.4612)(图23)。在298k下,配合物1-3的辐射衰减速率(kr)为7.5
×
104~2.4
×
105s-1
,发光寿命为1.2-2.3μs,比77k的寿命(139~818μs)小了2个数量级,表明1-3发光为热激活延迟荧光(tadf)。77k下,配合物1-3的最大发射波长为569,585和606nm(激发波长为365nm),与室温下的最大发射波长相比,发射谱带发生较弱的红移,这是因为低温下较低能级的激发态(t1)占主导。表3为采用自然键轨道(nbo)计算和分析得到的配合物1
–
3的单线态和三线态能级及δe(s
1-t1)。配合物1
–
3的s1和t1能级差分别为0.0723,0.0898和0.1067ev,能级差小于0.37ev,为证明配合物1
–
3具有tadf效应提供了进一步的证据。
[0092]
基于这些配合物的优化后的s1几何构型及homo和lumo的前线轨道图,还使用tddft计算了发射特性。计算结果表明了配合物1
–
3的发光主要来源于lumo到homo的电子跃迁。s1态下的lumo和homo前线轨道图如图24所示。由图知配合物homo中的电子主要集中在cu、卤素和p上,lumo中的电子主要集中在配体中,这说明了配合物1
–
3的发光主要来自xlct(卤素到配体的电荷跃迁)、mlct(金属到配体的电荷跃迁)和ilct配体内电荷跃迁(配体内电荷跃迁)。
[0093]
通过tddft计算优化的s0、s1和t1中cu中心的配位构型如图25所示。在s0、s1和t1构型中,双p配体与铜中心的键角(p-cu-p)变化很小,这归因于双膦配体dcdp的刚性。由键长键角变化可以看出由s0构型至s1和t1构型,配合物1的形变最小,故而配合物1拥有比配合物2和3更高的量子效率。以铜为中心的配合物1-3的s1和t1构型键角之和分别比s0构型键角之和变化了(1:-0.32~1.87
°
,0.79~2.65
°
;2:2.06~5.45
°
,-16.31~2.98
°
;3:-6.39~10.79
°
,-2.43~-5.87
°
)。变化较大的键角是p-cu-x,x-cu-x和p3-cu-x,导致了激发时的jahn-teller畸变。
[0094]
表6.配合物1-3在固态下的光物理数据.
[0095][0096][0097]a最大发射峰波长.
[0098]b发光寿命,实验误差
±
5%.
[0099]c空气中绝对量子效率,实验误差
±
5%.
[0100]d辐射衰减速率常数,kr=ф/τ
[0101]
e tddft计算垂直激发得到的能量(s1和t1能级,及s1和t1之间的能级差)
[0102]
本发明所提供的配合物1-3优化的构型(s0、s1和t1态)如图26所示。
[0103]
本发明所提供的配合物1-3的室温发光寿命(a:1;b:2;c:3)如图27所示。
[0104]
本发明提供了一种新型刚性咔唑苯基双膦配体及三个卤化亚铜配合物。配合物1、
2和3在室温固态下发绿黄光至黄橙光,最大发射波长分别为567,580和602nm。室温空气下固态绝对内量子效率φ
pl
为0.09~0.53,发光寿命为1.2-2.3μs,配合物1-3的发光为热激活延迟荧光,主要来自xlct(卤素到配体的电荷跃迁)、mlct(金属到配体的电荷跃迁)和ilct配体内电荷跃迁(配体内电荷跃迁),配体dcdp与3个配合物未见报道。配合物的发光寿命是目前报道的双核卤化亚铜配合物中寿命最低的。低的发光寿命有利于减小高浓度下光激子的猝灭效应,提升oled外量子效率。低寿命、高效发光配合物作为发光材料可应用于oled器件。
[0105]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1