一种罗贝考昔中间体及罗贝考昔的合成方法与流程
1.本发明属于药物合成技术领域,涉及一种非甾体抗炎药的合成方法,尤其是涉及一种罗贝考昔中间体及罗贝考昔的合成方法。
背景技术:
2.罗贝考昔(cas号:220991-32-2,英文名:robenacoxib),又称2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸,是一种新型的非甾体抗炎药,具有多种优异性能,包括起效快速以及可用于注射剂、口服制剂,可用于猫狗的炎症、疼痛和体温升高。与非选择性非甾体抗炎药相比,其对cox-2的抑制具有高度选择性,在猫血脑屏障和狗血脑屏障中起效快,安全性更高。
3.现有罗贝考昔的合成方法步骤长且使用大量的路易酸催化剂,污染大,收率低,同时反应过程要求无水条件,给工业化生产过程中的安全性和操作的便利性带来一定的困难。
4.文献报道罗贝考昔的合成主要有二类路线:
5.第一类合成路线,如,中国发明申请中公开一种酸的制备方法(公开号:cn109694330a),该方法以n-(4-乙基苯基)-2,3,5,6-四氟苯胺为原料与草酰氯经两次酰化反应得到中间体5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,再经水解、还原两步反应得到罗贝考昔,反应需要4步。反应方程式如下:
[0006][0007]
反应中需要大量的三氯化铝且是无水反应,操作不方便。
[0008]
第二类合成路线,如,公告号为cn1140500c的中国发明专利、公开号为cn112679410a的中国发明专利申请、公告号为cn109503399b的中国发明专利、公告号为cn102311355b的中国发明专利和公开号为cn107721901a的中国发明专利申请,以n-(4-乙基苯基)-2,3,5,6-四氟苯胺为原料与氯乙酰氯经两次酰化反应得到中间体5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2-酮,再经水解反应得到罗贝考昔。反应方程式如下:
[0009]
反应中需要大量的三氯化铝且是无水反应,操作不方便。
[0010]
又如,公开号为cn111807978a的中国发明专利申请提供了一种罗贝考昔的制备方法,该制备方法中,中间体5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2-酮的合成是以吲哚酮为原料,经酰化、还原、n-烷基化得到的,5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2-酮经进一步水解,即获得罗贝考昔。与前述方法相比,该方法只是中间体的合成方式不同,但仍大量使用三氯化铝,且还原反应时的还原试剂特殊,反应也要求无水条件,条件较为苛刻。
[0011][0012]
因此,亟需一种易于工业化生产的罗贝考昔的合成方法。
技术实现要素:
[0013]
本发明的目的是针对上述问题,提供了一种罗贝考昔中间体及罗贝考昔的合成方法。
[0014]
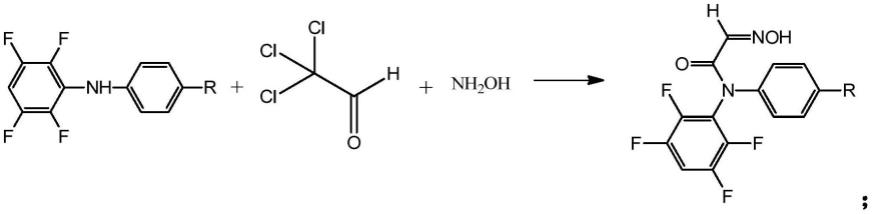
本发明创造性地提出了一种罗贝考昔中间体的合成方法,该合成方法以为原料,将该原料与三氯乙醛、羟胺经桑德迈尔反应合成获得所述的罗贝考昔中间体;该罗贝考昔中间体的化学式如下:
[0015]
其中,r为乙基或乙酰基。
[0016]
本发明采用桑德迈尔反应制备罗贝考昔中间体,整个反应过程中不使用三氯化铝等有毒、刺激性的原料,也不要求无水条件,具有污染少、易生产的优点,对反应条件要求低,能够显著降低工业化生产成本。
[0017]
进一步地,所述的桑德迈尔反应包括:
[0018]
a、原料与三氯乙醛、羟胺在酸性条件下进行第一步反应得到初级中间体,反应方程式如下:
[0019][0020]
b、再在酸催化下进行第二步反应得到罗贝考昔中间体,反应方程式如下:
[0021][0022]
即,原料为1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮或n-(4-乙基苯基)-2,3,5,6-四氟苯胺;初级中间体为n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺或n-(4-乙基苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺;罗贝考昔中间体为5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮或5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮。
[0023]
在上述的一种罗贝考昔中间体的合成方法中,步骤a中,原料、三氯乙醛和羟胺的摩尔比为0.66~1:1:1。
[0024]
在上述的一种罗贝考昔中间体的合成方法中,三氯乙醛为水合三氯乙醛或无水三氯乙醛,羟胺为盐酸羟胺或硫酸羟胺。
[0025]
在上述的一种罗贝考昔中间体的合成方法中,步骤a具体为:在第一容器中加入水、原料,搅拌下加入盐酸溶液或硫酸溶液,以提供酸性条件,备用;
[0026]
在第二容器中加入水、无水硫酸钠,升温至70~80℃后加入三氯乙醛,搅拌保温0.5小时,将上述第一容器中混合物加入第二容器中,再加入羟胺,升温80~85℃反应2小时后,降温至20℃,抽滤,水洗2次,真空干燥,得到初级中间体。
[0027]
在上述的一种罗贝考昔中间体的合成方法中,步骤b中,所述酸催化反应采用90%~98%硫酸溶液或多聚磷酸,90%~98%硫酸溶液或多聚磷酸的用量为初级中间体的2.5~5倍;反应温度为50~230℃,保温反应后冷却至室温,水洗,真空干燥得到罗贝考昔中间体。
[0028]
优选地,90%~98%硫酸溶液或多聚磷酸的用量为初级中间体的2.85-3.8倍。
[0029]
上述的一种罗贝考昔中间体的合成方法中,步骤b具体为:在第三容器中加入90%~98%硫酸溶液或多聚磷酸,以及初级中间体,搅拌下缓慢升温至50~230℃,保温反应2h;反应液冷却至室温,加入水,搅拌30min,抽滤,水洗,真空干燥后得到罗贝考昔中间体。
[0030]
本发明还提供了一种罗贝考昔的合成方法,该合成方法包括以下步骤:
[0031]
1)采用权利要求1-6所述的罗贝考昔中间体的合成方法,制备罗贝考昔中间体;
[0032]
2)罗贝考昔中间体经还原即得到罗贝考昔。
[0033]
作为优选,在上述的一种罗贝考昔的合成方法中,
[0034]
所述的罗贝考昔中间体的化学式为:即r为乙酰基,原料为1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮。
[0035]
则,上述罗贝考昔的合成方法中,步骤1)中,1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮与三氯乙醛、羟胺经桑德迈尔反应得到5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮;
[0036]
步骤2)中,5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮经还原后调酸得到罗贝考昔。
[0037]
如此,还原过程中,罗贝考昔中间体乙酰基上的羰基还原和二氢吲哚2,3号位上的羰基还原一步完成,这就使得本发明可以直接采用1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮作为原料,省略了将1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮还原为n-(4-乙基苯基)-2,3,5,6-四氟苯胺的步骤,提高了罗贝考昔的总收率(收率可达95%)。
[0038]
进一步,罗贝考昔的合成方法包括以下步骤:
[0039]
1)罗贝考昔中间体的合成
[0040]
a.1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮与三氯乙醛、羟胺在酸性条件下反应得到n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,反应方程式如下:
[0041][0042]
b.n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺在一定温度下通过酸催化反应得到5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,反应方程式如下:
[0043][0044]
2)罗贝考昔的合成
[0045]
5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮在碱性条件下经水合肼还原后调酸得到罗贝考昔,反应方程式如下:
[0046]
在上述的一种罗贝考昔的合成方法中,步骤2)中,罗贝考昔中间体在碱性条件下与二乙二醇经水合肼升温蒸馏除水,保温反应后冷却至室温,调酸至ph为2.9~3.1,搅拌,抽滤,水洗,真空干燥得到罗贝考昔。
[0047]
进一步,对于每100g罗贝考昔中间体,加入300~550ml二乙二醇和35~70g85%水合肼进行反应。进一步,上述步骤3)中所述的碱性条件是指ph为12~14。
[0048]
上述的一种罗贝考昔的合成方法中,步骤2)具体为:在第四容器中加入二乙二醇、罗贝考昔中间体、碱以及85%水合肼,搅拌下升温蒸馏除水至反应温度为200℃,保温反应4h;反应液冷却至室温,加入水,用盐酸溶液或硫酸溶液调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到罗贝考昔。
[0049]
上述的碱可以采用氢氧化钠或氢氧化钾。
[0050]
与现有技术相比,本发明的优点在于:
[0051]
1)本发明采用桑德迈尔反应制备罗贝考昔中间体,整个反应过程中不使用三氯化铝等有毒、刺激性的原料,也不要求无水条件,具有污染少、易生产的优点,对反应条件要求低,能够显著降低工业化生产成本;而后将罗贝考昔中间体还原即获得罗贝考昔。
[0052]
2)本发明罗贝考昔中间体乙酰基上的羰基还原和二氢吲哚2,3号位上的羰基还原一步完成,这就使得本发明可以直接采用1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮作为原料,省略了将1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮还原为n-(4-乙基苯基)-2,3,5,6-四氟苯胺的步骤,提高了罗贝考昔的总收率。
具体实施方式
[0053]
通过以下具体实施例进一步阐述;
[0054]
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其它不同于在此描述的其它方式来实施,因此本发明不受下面公开的具体实施例的限制。
[0055]
实施例1
[0056]
在250ml烧杯中加入100g水,56.6g(0.2mol)1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮,搅拌下加入25g 30%盐酸,备用。
[0057]
在1000ml三颈瓶中加入600ml水,170.4g(1.2mol)无水硫酸钠,升温至80℃后加入49.6g(0.3mol)水合三氯乙醛,搅拌保温0.5小时,将上述烧杯中混合物加入三颈瓶中,再加入20.9g(0.3mol)盐酸羟胺,升温至85℃反应2小时,降温至20℃,抽滤,水洗2次,真空干燥,
得到n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺70.2g,收率99%。
[0058]
在500ml三颈瓶中加入250g多聚磷酸,70.2gn-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,搅拌下缓慢升温至230℃,保温反应2h。反应液冷却至室温,加入200ml水,搅拌30min,抽滤,水洗,真空干燥后得到66.1g5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,收率99%。
[0059]
在500ml三颈瓶中加入250ml二乙二醇,66.1g5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,48.0g氢氧化钠,45.2g85%水合肼,搅拌下升温蒸馏除水至反应温度为200℃,保温反应4h。反应液冷却至室温,加入200ml水,30%盐酸调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸,收率95%(以1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮计)。
[0060]
实施例2
[0061]
在250ml烧杯中加入150g水,85.0g(0.3mol)1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮,搅拌下滴加入30g 98%硫酸,备用。
[0062]
在1000ml三颈瓶中加入700ml水,142.0g(1.0mol)无水硫酸钠,升温至70℃后加入49.6g(0.3mol)水合三氯乙醛,搅拌保温0.5小时,将上述烧杯中混合物加入三颈瓶中,再加入39.3g(0.3mol)硫酸羟胺,升温至80℃反应3小时,降温至20℃,抽滤,水洗2次,真空干燥,得到n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺105.2g,收率99%。
[0063]
在500ml三颈瓶中加入300g多聚磷酸,105.2gn-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,搅拌下缓慢升温至180℃,保温反应5h。反应液冷却至室温,加入200ml水,搅拌30min,抽滤,水洗,真空干燥后得到101.2g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,收率99%。
[0064]
在1000ml三颈瓶中加入400ml二乙二醇,101.2g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,84.0g氢氧化钾,67.1g85%水合肼,搅拌下升温蒸馏除水至反应温度为200℃,保温反应4h。反应液冷却至室温,加入400ml水,滴加98%硫酸调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸87.4g,收率89%(以1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮计)。
[0065]
实施例3
[0066]
在250ml烧杯中加入50g水,28.3g(0.1mol)1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮,搅拌下滴加入19.6g 98%硫酸,备用。
[0067]
在500ml三颈瓶中加入200ml水,14.2g(0.2mol)无水硫酸钠,升温至75℃后加入16.5g(0.1mol)水合三氯乙醛,搅拌保温0.5小时,将上述烧杯中混合物加入三颈瓶中,再加入13.1g(0.1mol)硫酸羟胺,升温至85℃反应2小时,降温至20℃,抽滤,水洗2次,真空干燥,得到n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺32.6g,收率92%。
[0068]
在500ml三颈瓶中加入100g 98%硫酸,32.6gn-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,搅拌下缓慢升温至50℃,保温反应6h。反应液冷却至室温,倾入200ml水中,搅拌30min,抽滤,水洗,真空干燥后得到29.5g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,收率95%。
[0069]
在500ml三颈瓶中加入100ml二乙二醇,29.5g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,12.0g氢氧化钠,11.3g85%水合肼,搅拌下升温蒸馏除水至反应温度
为200℃,保温反应4h。反应液冷却至室温,加入200ml水,30%盐酸调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸26.2g,收率80%(以1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮计)。
[0070]
实施例4
[0071]
在250ml烧杯中加入150g水,85.0g(0.3mol)1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮,搅拌下滴加入37.0g 30%盐酸,备用。
[0072]
在1000ml三颈瓶中加入500ml水、127.8g(0.9mol)无水硫酸钠,升温至70℃后加入57.9g(0.35mol)水合三氯乙醛,搅拌保温0.5小时,将上述烧杯中混合物加入三颈瓶中,再加入24.3g(0.35mol)盐酸羟胺,升温至80℃反应3小时,降温至20℃,抽滤,水洗2次,真空干燥,得到n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺105.2g,收率99%。
[0073]
在500ml三颈瓶中加入400g 90%硫酸,105.2gn-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,搅拌下缓慢升温至105℃,保温反应5h。反应液冷却至室温,倾入400ml水,搅拌30min,抽滤,水洗,真空干燥后得到95.1g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,收率95%。
[0074]
在1000ml三颈瓶中加入500ml二乙二醇,95.1g 5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,67.2g氢氧化钾,45.2g85%水合肼,搅拌下升温蒸馏除水至反应温度为200℃,保温反应4h。反应液冷却至室温,加入400ml水,滴加98%硫酸调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸83.5g,收率85%(以1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮计)。
[0075]
实施例5
[0076]
在1l烧杯中加入300g水,134.6g(0.5mol)n-(4-乙基苯基)-2,3,5,6-四氟苯胺,搅拌下滴加入60.8g 30%盐酸,备用。
[0077]
在2l三颈瓶中加入1l水、284.0g(2.0mol)无水硫酸钠,升温至70℃后加入99.3g(0.6mol)水合三氯乙醛,搅拌保温0.5小时,将上述烧杯中混合物加入三颈瓶中,再加入41.7g(0.6mol)盐酸羟胺,升温至80℃反应3小时,降温至20℃,抽滤,水洗2次,真空干燥,得到n-(4-乙基苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺168.4g,收率99%。
[0078]
在1l三颈瓶中加入700g 90%硫酸,202.1gn-(4-乙基苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺,搅拌下缓慢升温至100℃,保温反应5h。反应液冷却至室温,倾入1l水,搅拌30min,抽滤,水洗,真空干燥后得到155.2g 5-乙基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,收率97%。
[0079]
在2l三颈瓶中加入700ml二乙二醇,155.2g 5-乙基基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮,48.0g氢氧化钠,37.7g85%水合肼,搅拌下升温蒸馏除水至反应温度为200℃,保温反应4h。反应液冷却至室温,加入700ml水,滴加98%硫酸调ph为3,搅拌30min,抽滤,水洗,真空干燥后得到2-(5-乙基-2-((2,3,5,6-四氟苯基)氨基)苯基)乙酸144.0g,收率88%(以n-(4-乙基苯基)-2,3,5,6-四氟苯胺计)。
[0080]
本文中所描述的具体实施例仅仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
[0081]
尽管本文较多地使用了中1-(4-((2,3,5,6-四氟苯基)氨基)苯基)乙烷-1-酮、n-(4-乙酰苯基)-2-(羟基亚氨基)-n-(2,3,5,6-四氟苯基)乙酰胺、5-乙酰基-1-(2,3,5,6-四氟苯基)二氢吲哚-2,3-二酮、罗贝考昔、三氯乙醛、羟胺、桑德迈尔反应等术语。使用这些术语仅仅是为了更方便地描述和解释本发明的本质,把它们解释成任何一种附加的限制都是与本发明精神相违背的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1