海洋曲霉真菌及其用途和分析方法
1.本发明属于海洋生物技术领域,具体涉及海洋曲霉真菌。
背景技术:
2.海洋生态是巨大的天然资源宝库,其内生存的微生物数目占地球微生物的绝大部分,每一种生活其中的生物都展现着海洋与陆地不同的特殊性。与陆生生物不同,海洋的研究发展史比陆地较短。经大量研究发现,海洋微生物的次代谢产物种类繁多,且多具有生物活性。并且海洋微生物受其自身结构、生理和遗传等特点,繁殖周期短较短,因此采用微生物发酵法制备各种活性代谢产物,受外界影响因素较少,培养条件易于控制,提取方法便于操作。
3.神经酰胺是一种具有多种生物学功能活性的天然化合物,但由于其价格昂贵,目前在某些领域中对它的研究依然涉及较少。在部分海洋微生物的次代谢产物中有发现神经酰胺的报道。但截至目前还未有一种能大量产生神经酰胺的微生物,且目前针对这类微生物及其活性产物缺少相应的分析评价方法。这不利于海洋微生物及神经酰胺等活性产物的研究和利用。
技术实现要素:
4.本发明的目的在于克服现有技术的不足之处,提供了海洋曲霉真菌及其用途和分析方法。
5.本发明解决其技术问题所采用的技术方案之一是:
6.一种海洋曲霉真菌aspergillus sp.ds142-1,保藏于广东省微生物菌种保藏中心(gdmcc),保藏地址为广州市先烈中路100号大院59号楼5楼,保藏日期为2021年09月14日,保藏编号为gdmcc no:61933。
7.本发明解决其技术问题所采用的技术方案之二是:
8.一种海洋曲霉真菌aspergillus sp.ds142-1在制备神经酰胺中的用途。
9.本发明解决其技术问题所采用的技术方案之三是:
10.一种海洋曲霉真菌aspergillus sp.ds142-1中的神经酰胺的分析方法,包括:
11.1)所述的海洋曲霉真菌aspergillus sp.ds142-1的发酵物用乙酸乙酯处理得到粗产物;
12.2)所述粗产物用c
18
固相萃取进行纯化:将拌好硅藻土的所述粗产物装进活化后的c
18
固相萃取柱中,先用5~6个柱体积的94~96%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并以100%甲醇的洗脱成分作为纯化产物;
13.3)所述纯化产物用hplc进行神经酰胺含量测定:色谱条件:ultimate xb-c18色谱柱:4.6
×
250mm,5μm;流动相超纯水-甲醇,流速0.7~0.9ml/min;检测波长210nm;进样量7~9μl;柱温25~27℃;100%甲醇洗脱15~20min。
14.进一步地,步骤1)中,发酵物的制备方法包括:
15.a)所述的海洋曲霉真菌aspergillus sp.ds142-1复苏后,涂布于pda培养基,24~26℃下培养70~75h进行活化,随后接种于pdb液体培养基,培养基盐度为14~16ppt,ph值为7.2~7.4,温度为24~26℃,振摇转速为175~185r
·
min-1
,发酵时间为22~26h,培养得到种子液;
16.b)取所述种子液接种于发酵培养基,ph值为7.2~7.4,温度为24~26℃,振摇转速为145~155r
·
min-1
,发酵时间为70~75h,培养得到所述发酵物。
17.进一步地,所述发酵培养基的配方包括:可溶性淀粉44~46g/l,酵母提取粉7~9g/l,盐度为14~16ppt,ph值为7.2~7.4。
18.进一步地,步骤1)中,用乙酸乙酯处理的方法包括:所述发酵物过滤分离得到发酵液与菌丝体;所述发酵液用乙酸乙酯萃取得到第一乙酸乙酯萃取液;所述菌丝体用乙酸乙酯-甲醇-乙酸混合液提取、过滤,滤液用乙酸乙酯萃取得到第二乙酸乙酯萃取液,反复提取、萃取若干次;合并第一乙酸乙酯萃取液和第二乙酸乙酯萃取液,除去溶剂后用甲醇复溶,得到粗产物。
19.进一步地,所述乙酸乙酯-甲醇-乙酸混合液中,乙酸乙酯、甲醇和乙酸的体积比为75~85:14~16:4~6。
20.进一步地,步骤2)中,所述c
18
固相萃取柱的活化方法包括:按水
→
29~31%甲醇
→
59~61%甲醇
→
94~96%甲醇
→
100%甲醇
→
94~96%甲醇的顺序依次洗脱活化c
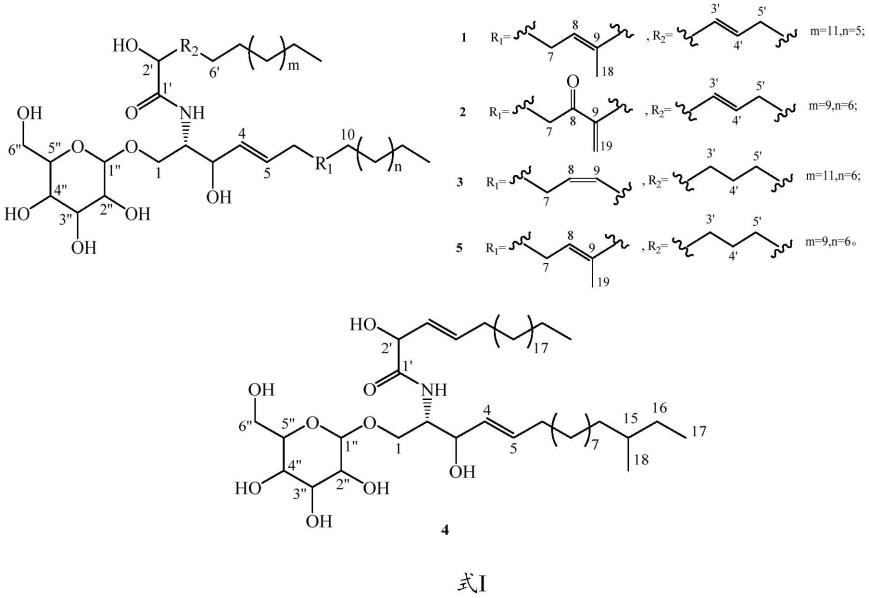
18
固相萃取柱,每个体系洗脱量为4~5个柱体积。
21.本发明所涉及的设备、试剂、工艺、参数等,除有特别说明外,均为常规设备、试剂、工艺、参数等,不再作实施例。
22.本发明所列举的所有范围包括该范围内的所有点值。
23.本发明中,%在表示浓度时,除有特别说明或在领域内有通用意义外,溶质为液体时%代表体积百分比,溶质为固体时%代表g/100ml。
24.本技术方案与背景技术相比,它具有如下优点:
25.本发明从福建省漳州市东山湾海域的珊瑚共附生微生物中分离得到一种海洋曲霉真菌aspergillus sp.ds142-1 gdmcc no:61933。该海洋曲霉真菌可产生多种神经酰胺化合物,具有潜在的应用前景。同时,本发明建立了该海洋曲霉真菌发酵物中神经酰胺的分离纯化和定量分析方法,为该种海洋微生物及其神经酰胺等活性产物的研究和利用提供了思路。
附图说明
26.图1为本发明实施例中海洋曲霉真菌aspergillus sp.ds142-1的发酵物的提取流程示意图。
27.图2为本发明实施例中c
18
固相萃取采用75%甲醇洗脱后的hplc图谱。
28.图3为本发明实施例中c
18
固相萃取采用80%甲醇洗脱后的hplc图谱。
29.图4为本发明实施例中c
18
固相萃取采用85%甲醇洗脱后的hplc图谱。
30.图5为本发明实施例中c
18
固相萃取采用90%甲醇洗脱后的hplc图谱。
31.图6为本发明实施例中c
18
固相萃取采用95%甲醇洗脱后的hplc图谱。
32.图7为本发明实施例中的神经酰胺标准曲线。
33.图8为式ⅱ所示神经酰胺化合物ds142-1-18-2-3-3-6即神经酰胺标准品的hplc图谱。
具体实施方式
34.下面结合附图和实施例对本发明作进一步说明。
35.实施例
36.1.菌种
37.本实施例的海洋曲霉真菌aspergillus sp.ds142-1,分离自福建省漳州市东山湾海域的珊瑚共附生微生物中,菌株现保藏于广东省微生物菌种保藏中心(gdmcc),保藏地址为广州市先烈中路100号大院59号楼5楼,保藏日期为2021年09月14日,保藏编号为gdmcc no:61933。
38.2.培养基
39.固体培养基(pda):马铃薯(从中提取浸出粉)300.0g,葡萄糖20.0g,琼脂15.0g,氯霉素0.1g,适量生态矿物盐溶于纯水(初始盐度为15ppt),初始ph值7.2~7.4(用3mol/l hcl和6mol/l naoh调节),高压锅121℃灭菌2h。
40.液体培养基(pdb):马铃薯(从中提取浸出粉)300.0g,葡萄糖20.0g,适量生态矿物盐溶于纯水(初始盐度为15ppt),初始ph值7.2~7.4(用3mol/l hcl和6mol/l naoh调节),高压锅121℃灭菌2h。
41.发酵培养基:可溶性淀粉45g/l,酵母提取粉8g/l,适量生态矿物盐溶于纯水(初始盐度为15ppt),初始ph值7.2~7.4(用3mol/l hcl和6mol/l naoh调节),高压锅121℃灭菌2h。
42.3.培养与发酵条件
43.1)将冻存于-80℃超低温冰箱中的菌种悬液梯度溶解后,在超净工作台内取少量菌液置于含有凝固pda培养基的平板上进行涂布活化,25℃下培养72h后,蘸取经过活化后的菌株孢子,接种于250ml三角锥形瓶中,该瓶中装有100ml pdb液体培养基,于光照摇床培养箱中摇瓶培养24h后得到种子液。发酵条件:培养基盐度:15ppt;ph:7.2~7.4;温度:25℃;摇床转速:180r
·
min-1
;发酵时间:24h。
44.2)用移液器移取2ml种子液于100ml发酵培养基内,培养72h后得到发酵物。发酵条件:ph:7.2~7.4;温度:25℃;摇床转速:150r
·
min-1
;发酵时间:72h。
45.4.样品提取
46.发酵物用四层纱布过滤,分离得到菌丝体与发酵液。发酵液转移至分液漏斗内,加入一定量乙酸乙酯振荡萃取并静置稍许,分离取上层液,为乙酸乙酯萃取液;菌丝体用80:15:5(v/v/v)的乙酸乙酯-甲醇-乙酸混合液60ml,于超声波清洗机内提取40min后,用四层纱布过滤出菌丝体,滤液依旧加入一定量乙酸乙酯振荡,再加入适量纯水使之分层,充分振荡萃取后静置稍许,使之分层,分离取上层液,为乙酸乙酯萃取液;以上步骤反复提取和萃取2~3次。以上每次分离收集得的乙酸乙酯萃取液于同一已知重量的锥形瓶内旋蒸。用1.5ml甲醇超声波溶解提取旋蒸所得的粗产物。
47.该菌发酵得到的粗产物中可分离得到多种类的神经酰胺(如式ⅰ所示),本实施例针对从中分离得到的其中一个带糖基的脑苷脂神经酰胺(如式ⅱ所示)进行纯化和分析。
[0048][0049][0050]
5.含有神经酰胺的粗产物的纯化
[0051]
按上述操作步骤获得的粗产物,成分复杂,hplc图像中杂质峰多,不利于神经酰胺的定性定量。所以使用c
18
固相萃取对乙酸乙酯萃取物进行初步分离,方便后续的hplc检测。
[0052]
甲醇溶解的粗产物用1.5倍重的硅藻土拌样备用。分别使用不同浓度的甲醇进行洗脱,除去粗产物中的杂质化合物。对用不同浓度甲醇进行洗脱除杂后得到的分离物分别进行hplc检测,比较选择适合的c
18
固相萃取分离洗脱条件。具体如下:
[0053]c18
固相萃取条件1(75%甲醇洗脱):
[0054]
按超纯水
→
25%甲醇
→
50%甲醇
→
75%甲醇
→
100%甲醇
→
75%甲醇的顺序依次洗脱活化c
18
固相萃取柱(nanochrom),每个体系洗脱量为4~5个柱体积。活化后将拌好硅藻土的样品装进c
18
固相萃取柱中,先用5~6个柱体积的75%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并保留100%甲醇的洗脱成分,将其旋蒸后使用1ml甲醇超声溶解,再通过0.22微米滤膜过滤得到样品,最后进行hplc分析(图2)。
[0055]c18
固相萃取条件2(80%甲醇洗脱):
[0056]
按超纯水
→
20%甲醇
→
40%甲醇
→
60%甲醇
→
80%甲醇
→
100%甲醇
→
80%甲醇的顺序依次洗脱活化c
18
固相萃取柱,每个体系洗脱量为4~5个柱体积。活化后将拌好硅藻
土的样品装进c
18
固相萃取柱中,先用5~6个柱体积的80%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并保留100%甲醇的洗脱成分,将其旋蒸后使用1ml甲醇超声溶解,再通过0.22微米滤膜过滤得到样品,最后进行hplc分析(图3)。
[0057]c18
固相萃取条件3(85%甲醇洗脱):
[0058]
按超纯水
→
30%甲醇
→
60%甲醇
→
85%甲醇
→
100%甲醇
→
85%甲醇的顺序依次洗脱活化c
18
固相萃取柱,每个体系洗脱量为4~5个柱体积。活化后将拌好硅藻土的样品装进c
18
固相萃取柱中,先用5~6个柱体积的85%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并保留100%甲醇的洗脱成分,将其旋蒸后使用1ml甲醇超声溶解,再通过0.22微米滤膜过滤得到样品,最后进行hplc分析(图4)。
[0059]c18
固相萃取条件4(90%甲醇洗脱):
[0060]
按超纯水
→
30%甲醇
→
60%甲醇
→
90%甲醇
→
100%甲醇
→
90%甲醇的顺序依次洗脱活化c
18
固相萃取柱,每个体系洗脱量为4~5个柱体积。活化后将拌好硅藻土的样品装进c
18
固相萃取柱中,先用5~6个柱体积的90%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并保留100%甲醇的洗脱成分,将其旋蒸后使用1ml甲醇超声溶解,再通过0.22微米滤膜过滤得到样品,最后进行hplc分析(图5)。
[0061]c18
固相萃取条件5(95%甲醇洗脱):
[0062]
按超纯水
→
30%甲醇
→
60%甲醇
→
95%甲醇
→
100%甲醇
→
95%甲醇的顺序依次洗脱活化c
18
固相萃取柱,每个体系洗脱量为4~5个柱体积。活化后将拌好硅藻土的样品装进c
18
固相萃取柱中,先用5~6个柱体积的95%甲醇洗脱除杂,再用5~6柱体积的100%甲醇进行洗脱,并保留100%甲醇的洗脱成分,将其旋蒸后使用1ml甲醇超声溶解,再通过0.22微米滤膜过滤得到样品,最后进行hplc分析(图6)。
[0063]
hplc检测条件:ultimate xb-c18色谱柱:4.6
×
250mm,5μm;流动相超纯水-甲醇,流速0.8ml/min;检测波长210nm;进样量8μl;温度26℃;100%甲醇洗脱20min。
[0064]
根据210nm波长检测可知,神经酰胺的出峰时间为16~17min。以上hplc结果对比可以看出,c
18
固相萃取柱使用95%甲醇去除杂质后,得到的分离物进行hplc检测杂质峰最少,因此选择该条件作为c
18
固相萃取的洗脱条件。
[0065]
6.神经酰胺含量的测定
[0066]
粗产物通过上述优化后的c
18
固相萃取条件进行纯化,随后用hplc进行神经酰胺含量测定:
[0067]
1)色谱检测条件:ultimate xb-c
18
色谱柱:4.6
×
250mm,5μm;流动相超纯水-甲醇,流速0.8ml/min;检测波长210nm;进样量8μl;柱温26℃;100%甲醇洗脱20min。
[0068]
2)神经酰胺标准曲线的制作:
[0069]
将乙酸乙酯层浓缩物(即上述粗产物,记为ds142-1)过正相硅胶柱(200~300目),过柱条件为:100%石油醚
→
石油醚∶乙酸乙酯=90∶1
→
石油醚∶乙酸乙酯=50∶1
→
石油醚∶乙酸乙酯=30∶1
→
石油醚∶乙酸乙酯=10∶1
→
石油醚∶乙酸乙酯=5∶1
→
石油醚∶乙酸乙酯=3∶1
→
石油醚∶乙酸乙酯=1∶1
→
100%石油醚
→
100%甲醇,得到18个组分。按出峰顺序取18个组分中的第2个组分ds142-1-18-2(2.1719g)过正相常压柱(300~400目硅胶),过柱条件为二氯甲烷∶甲醇=36∶1
→
二氯甲烷∶甲醇=32∶1
→
二氯甲烷∶甲醇=30∶1
→
二氯甲烷∶甲醇=20∶1
→
100%甲醇,得到5个组分,ds142-1-18-2-1(20.1mg),ds142-1-18-2-2
(17.5mg),ds142-1-18-2-3(1.0511g),ds142-1-18-2-4(200mg),ds142-1-18-2-5(880mg)。取ds142-1-18-2-3过正相常压柱(300~400目硅胶),过柱条件:100%二氯甲烷
→
二氯甲烷∶甲醇=36∶1
→
二氯甲烷∶甲醇=30∶1
→
二氯甲烷∶甲醇=20∶1
→
二氯甲烷∶甲醇=10∶1
→
二氯甲烷∶甲醇=5∶1
→
二氯甲烷∶甲醇=1∶1
→
100%甲醇,得到4个组分。取ds142-1-18-2-3-3(88.4mg)进行液相分析和制备(welchxb-c18柱s-5μm 10
×
250mm,100%甲醇,210nm,254nm,l:2ml/min),得到8个组分,ds142-1-18-2-3-3-1(2.3mg),ds142-1-18-2-3-3-2(3.5mg),ds142-1-18-2-3-3-3(2.0mg),ds142-1-18-2-3-3-4(1.5mg),ds142-1-18-2-3-3-5(4.7mg),ds142-1-18-2-3-3-6(14.2mg),ds142-1-18-2-3-3-7(2.7mg),ds142-1-18-2-3-3-8(3.5mg),如图8所示,经h、c谱和质谱可以鉴定为神经酰胺类化合物,将ds142-1-18-2-3-3-6即上述式ⅱ所示化合物作为神经酰胺标准品。
[0070]
将神经酰胺标准品称重,加甲醇配制成1000μg/ml的神经酰胺标准溶液。根据所设计的浓度依次加适量甲醇将1000μg/ml的神经酰胺标准溶液稀释成200μg/ml、100μg/ml、75μg/m、50μg/ml、25μg/ml和10μg/ml,具体的稀释步骤如下:
[0071]
200μg/ml:移液管移取1000μg/ml的标准液2ml于10ml容量瓶,加甲醇定容。
[0072]
100μg/ml:移液管移取1000μg/ml的标准液1ml于10ml容量瓶,加甲醇定容。
[0073]
75μg/m:移液管移取150μg/ml的标准液5ml于10ml容量瓶,加甲醇定容。
[0074]
50μg/ml:移液管移取100μg/ml的标准液5ml于10ml容量瓶,加甲醇定容。
[0075]
25μg/ml:移液管移取50μg/ml的标准液5ml于10ml容量瓶,加甲醇定容
[0076]
10μg/ml:移液管移取100μg/ml的标准液1ml于10ml容量瓶,加甲醇定容。
[0077]
以上神经酰胺标准溶液依次于hplc分析测定,每个浓度的神经酰胺标准溶液平行测定3次,选择210nm波长处理hplc结果,根据hplc上所示峰面积计算其与神经酰胺浓度之间的线性关系,拟合神经酰胺标准曲线(如图7)。
[0078]
3)样品溶液的配制:粗产物通过上述优化后的c
18
固相萃取条件进行纯化后,所得产物用一定量的甲醇超声溶解并定容至1.0ml,经尼龙膜过滤至样品分析瓶内备用。进样,检测。
[0079]
4)检测结果带入标准曲线中,根据hplc的峰面积结果计算提取所得的神经酰胺浓度,各试验因素下各水平的平行试验结果运用microsoft excel软件计算浓度均值,同一影响因素不同水平间的试验结果采用ibm spss statistics 21统计软件分析处理,比较显著性差异。采用origin 2019软件进行图表绘制。
[0080]
以上所述,仅为本发明较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1