一种具有窄谱带发射的芳基硼氮类化合物及其制备和应用
1.本发明涉及发光材料技术领域,更具体的,涉及一种具有窄谱带发射的芳基硼氮类化合物及其制备和应用。
背景技术:
2.有机发光二极管(organic light-emitting diodes,简称oled)是一种新型的发光器件,具有可弯曲、广视角、自发光、节能、驱动电压低、响应速度快以及可柔性加工等优点,成为当前产业化的热点趋势。oled在全彩色显示和照明应用技术中取得了较大的进展,oled是一个由多层有机功能层和电极组成的叠层结构。其中有机功能层包括注入层、传输层、阻挡层以及发光层等。发光层作为电致发光器件的核心,决定了器件的效率和光色,其材料的开发研宄得到了更加广泛的关注。
3.有机发光二极管的发光原理是对于有机发光二极管施加电压,注入来自阳极的空穴与来自阴极的电子,它们在发光层中再次结合形成激子。依据电子自旋统计法,单线态激子与三重态激子以25%:75%的比例生成。按照oled发光原理,可分为荧光型与磷光型两种类型。荧光型因为使用了单线态激子来发光,故其内部量子效率只能达到25%。热致延迟荧光(thermally-activated delayed fluorescence,tadf)材料是继有机荧光材料和有机磷光材料之后发展的第三代有机发光材料。tadf材料一般具有较小的单线态-三线态能级差(e
st
),三线态激子可以通过反间隙穿越转变成单线态激子发光,可以充分利用电激发下形成的单线态激子和三线态激子,器件的内量子效率可以达到100%,同时材料结构可控,性质稳定,价格便宜无需贵重金属,在oled领域的应用前景广阔。
4.现有技术在oled器件中引入窄光谱tadf材料作为发光染料,有机发光器件中发光层由主体材料3,3'-二(9h-咔唑-9-基)-1,1'-联苯(mcbp)和掺杂的客体tadf材料构成,现有技术tadf材料分子之间产生了严重的π-π堆积作用,使得tadf材料掺杂浓度小于发光层质量的10%,低浓度的掺杂所要求的真空蒸镀制备工艺条件要求更加苛刻,难以控制掺杂比例。而在发光材料中进行高浓度客体材料掺杂时,现有技术制备的发光材料易发生聚集诱导荧光淬灭的现象,导致发光材料荧光量子产率难以达到预期的效果,导致发光效率低。同时由于强烈的分子内电荷转移导致其色纯度很差,无法满足全彩显示技术高色纯度的技术需求,因此,能够实现高浓度掺杂且具有高发光效率、高色纯度的新型材料体系亟待开发。
技术实现要素:
5.本发明为克服现有技术所述的窄光谱tadf材料与主体材料在进行高比例掺杂时,易发生荧光淬灭现象,从而导致发光材料发光效率低的技术缺陷,提供一种能够与主体材料实现高比例掺杂,具有窄谱带发射的芳基硼氮类化合物。
6.本发明的另一目的在于提供所述具有窄谱带发射的芳基硼氮类化合物的制备方法。
7.本发明的另一目的在于提供所述具有窄谱带发射的芳基硼氮类化合物在制备发光材料中的应用。
8.本发明的另一目的在于提供所述具有窄谱带发射的芳基硼氮类化合物在制备发光器件中的应用。
9.为解决上述技术问题,本发明采用的技术方案是:
10.一种具有窄谱带发射的芳基硼氮类化合物,所述化合物化学结构式如式a1或式a2所示:
[0011][0012]
本发明提供的具有窄谱带发射的芳基硼氮类化合物以含叔丁基咔唑骨架的三配位硼发光化合物为中心,通过在分子骨架外围引入不同位置取代的大空间位阻的给电子基团——三苯胺,由于三苯胺上的苯环能够旋转,形成了扭曲的结构,从而抑制分子之间的π-π堆积作用,预防荧光淬灭,因而可以与主体材料进行高浓度掺杂的同时确保高的荧光量子产率。而类似三甲基苯基团、苯咔唑基团等取代基团的位阻小,并且其并不像三苯胺那样有能够自由旋转的基团,所以无法与主体材料形成高浓度掺杂。同时,三苯胺具有中等的给电子能力,当其引入到硼氮的分子骨架中不会产生强烈的分子内电荷转移,使得整个分子实现刚柔适度的共轭结构以降低分子的重组能,实现分子激发态下的结构形变小,获得窄带的发射即高色纯度,解决现有技术中有机发光二极管无法同时满足全彩显示技术高掺杂浓度、高效率以及高色纯度技术问题。
[0013]
本发明还保护一种具有窄谱带发射的芳基硼氮类化合物的制备方法如下:
[0014]
将含咔唑骨架三配位硼的化合物、4-溴-n,n-二苯基苯胺和/或3-溴-n,n-二苯基苯胺、四三苯基膦钯和碳酸钾配制成混合液,惰性气体下,将混合液加热得到所述具有窄谱带发射的芳基硼氮类化合物;所述含咔唑骨架三配位硼的化合物,其结构式为所述混合液内含咔唑骨架三配位硼的化合物、4-溴-n,n-二苯基苯胺和/或3-溴-n,n-二苯基苯胺、四三苯基膦钯和碳酸钾的摩尔比为:1:1~1.2:0.04~0.06:2~3;所述混合液中有机溶剂为四氢呋喃、4-甲基四氢呋喃、1,4-二氧六环的一种及以上;s4中所述加热温度为65~85℃,时间为12~16h。
[0015]
本发明所述的一种具有窄谱带发射的芳基硼氮类化合物还需进行后处理,具体方法如下:二氯甲烷萃取、无水硫酸钠干燥有机层、过滤蒸发、柱层析纯化、真空干燥,所述柱层析使用的洗脱剂为二氯甲烷:石油醚=1:10(v/v)。
[0016]
进一步的,所述含咔唑骨架三配位硼的化合物的制备方法如下:将4,4'-二叔丁
基-2,2'-二吡啶、1,5-环辛二烯氯化铱二聚体和/或甲氧基(环辛二烯)合铱二聚体、中间体化合物m2和联硼酸频那醇酯配置成混合溶液经加热反应得到所述的含咔唑骨架三配位硼的化合物;所述中间体化合物m2其结构式为所述混合液中4,4'-二叔丁基-2,2'-二吡啶、1,5-环辛二烯氯化铱二聚体和/或甲氧基(环辛二烯)合铱二聚体、中间体化合物m2和联硼酸频那醇酯的摩尔比为:0.02~0.03:0.01~0.02:1:1~2;所述混合液中有机溶剂选自四氢呋喃、4-甲基四氢呋喃、1,4-二氧六环的一种及以上;s3中所述加热温度为70~90℃,时间为12~16h。
[0017]
本发明所述含咔唑骨架三配位硼的化合物还需进行后处理,具体方法如下:二氯甲烷萃取、无水硫酸钠干燥有机层、减压蒸馏、柱层析纯化、真空干燥,所述柱层析使用的洗脱剂为二氯甲烷:石油醚=1:4(v/v)。
[0018]
更进一步的,所述中间体化合物m2的制备方法如下:将正丁基锂和/或叔丁基锂、中间体化合物m1,冷却到-35~-45℃,加入三溴化硼;在0℃下加入n,n-二异丙基乙胺,形成混合液,将混合液加热反应得到;所述中间体化合物m1其结构式为所述混合液中正丁基锂或叔丁基锂、中间体m1、三溴化硼、n,n-二异丙基乙胺的摩尔比为1~2:1:1~2:1~2;;所述加热温度为110~120℃,时间为12~16h。
[0019]
本发明所述中间体m1和中间体m2还需进行后处理,具体方法如下:冷却、二氯甲烷萃取、脱溶剂、柱层析纯化,所述柱层析使用的洗脱剂为石油醚。
[0020]
更进一步的,所述中间体化合物m1的制备方法如下:将3,6-二叔丁基咔唑、叔丁醇钾和/或叔丁醇钠、充分混合后加入2-溴-1,3-二氟苯,在惰性气体下,加热反应得到;所述3,6-二叔丁基咔唑、叔丁醇钾和/或叔丁醇钠、2-溴-1,3-二氟苯均是溶解在n,n二甲基甲酰胺、二甲基亚砜、n,n二甲基乙酰胺中的一种及以上,形成混合液;所述混合液中3,6-二叔丁基咔唑、叔丁醇钾或叔丁醇钠、2-溴-1,3-二氟苯的摩尔比为:2~2.5:2~3:1;所述加热温度为140~150℃,时间为24~36h。
[0021]
本发明所述的惰性气体选择氦气、氮气、氩气的一种及以上。
[0022]
本发明还保护所述具有窄谱带发射的芳基硼氮类化合物在制备发光材料中的应用。
[0023]
本发明所述的发光材料,包含具有窄谱带发射的芳基硼氮类化合物和主体材料。
[0024]
本发明所述主体材料为3,3'-二(9h-咔唑-9-基)-1,1'-联苯,即mcbp。
[0025]
本发明所述发光材料中具有窄谱带发射的芳基硼氮类化合物占发光材料质量的1%~30%。
[0026]
优选的,本发明所述发光材料中具有窄谱带发射的芳基硼氮类化合物占发光材料质量的10%~30%。
[0027]
本发明还保护所述具有窄谱带发射的芳基硼氮类化合物在制备发光器件中的应
用。
[0028]
本发明所述的发光器件包括发光层;所述发光层包括上述的发光材料。
[0029]
与现有技术相比,本发明的有益效果是:
[0030]
1.本发明提供的芳基硼氮类化合物发光效率高。本发明提供的具有窄谱带发射的芳基硼氮类化合物以含叔丁基咔唑骨架的三配位硼发光化合物为中心,通过在分子骨架外围引入不同位置取代的大空间位阻的给电子基团——三苯胺,由于三苯胺上的苯环能够旋转,形成了扭曲的结构,从而抑制分子之间的π-π堆积作用,预防荧光淬灭,因而可以与主体材料mcbp进行高浓度的掺杂。本发明所述化合物在主体材料中掺杂比例高达30%时,所制备的发光材料荧光量子产率达到87%,拥有更高的发光效率,远超同比例掺杂的其它化合物的荧光量子产率。
[0031]
2.本发明提供的芳基硼氮类化合物通过引入具有中等的给电子能力的三苯胺,当其引入到硼氮的分子骨架中不会产生强烈的分子内电荷转移,使得整个分子实现刚柔适度的共轭结构以降低分子的重组能,实现分子激发态下的结构形变小,获得窄带的发射。
附图说明
[0032]
图1是实施例1所得产物a1的核磁共振氢谱图。
[0033]
图2是实施例2所得产物a2的核磁共振氢谱图。
[0034]
图3是实施例1所得产物a1的质谱图。
[0035]
图4是实施例2所得产物a2的质谱图。
[0036]
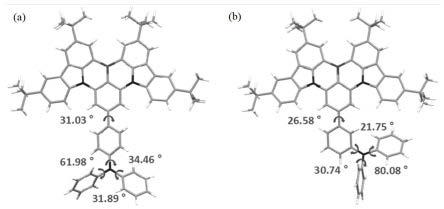
图5中(a)为实施例1制备得到的产物a1的晶体结构图;(b)为实施例2制备得到的产物a2的晶体结构图。
[0037]
图6为实施例1制备得到的产物a1的晶体分子间作用力结构图。
[0038]
图7为实施例2制备得到的产物a2的晶体分子间作用力结构图。
[0039]
图8是实施例1所得产物a1在甲苯溶液中的紫外可见吸收光谱图和荧光发射光谱图。
[0040]
图9是实施例2所得产物a2在甲苯溶液中的紫外可见吸收光谱图和荧光发射光谱图。
[0041]
图10是实施例1所得产物a1鼓入氮气前后荧光强度变化的荧光光谱图。
[0042]
图11是实施例2所得产物a2鼓入氮气前后荧光强度变化的荧光光谱图。
[0043]
图12是实施例1所得产物a1在甲苯溶液中常温荧光和低温磷光图。
[0044]
图13是实施例1所得产物a2在甲苯溶液中常温荧光和低温磷光图。
[0045]
图14是实施例1~2所得产物a1和a2的热重分析图。
[0046]
图15是实施例1~2所得产物a1和a2制成薄膜的荧光发射光谱图,其中(a)图是制备实施例1~2的荧光发射光谱图,(b)图是制备实施例3~4的荧光发射光谱图,(c)图是制备实施例5~6的荧光发射光谱图。
[0047]
图16是制备对比例1~4制成薄膜的荧光发射光谱图,其中(a)图是制备对比例1~2的荧光发射光谱图,(b)图是制备对比例3~4的荧光发射光谱图。
[0048]
图17是制备实施例1~6制成薄膜的荧光寿命图,其中(a)图是制备实施例1~2的荧光寿命图,(b)图是制备实施例3~4的荧光寿命图,(c)图是制备实施例5~6的荧光寿命
图。
[0049]
图18是制备对比例1~4制成薄膜的荧光寿命图,其中(a)图是制备对比例1~2的荧光寿命图,(b)图是制备对比例3~4的荧光寿命图。
[0050]
图19是实施例1~2所得产物a1和a2在甲苯溶液中的温度依赖的荧光寿命图,其中(a)制备实施例1温度依赖的荧光寿命图,其中(b)制备实施例2温度依赖的荧光寿命图。
具体实施方式
[0051]
下面结合具体实施方式对本发明作进一步的说明,但实施例并不对本发明做任何形式的限定。本领域技术人员在理解本发明的基础上对本发明所进行的变更、替换、改进依旧属于本发明的保护范围。除非另有说明,本发明实施例采用的原料试剂为常规购买的原料试剂。
[0052]
原料来源:天津市大茂化学试剂厂、广州化学试剂厂、安徽泽升科技有限公司、上海毕得医药科技股份有限公司
[0053]
实施例1
[0054]
一种具有窄谱带发射的芳基硼氮类化合物,化合物化学结构式如a1
[0055][0056]
具有窄谱带发射的芳基硼氮类化合物的制备方法,包括以下步骤:
[0057]
s1.将含有3,6-二叔丁基-9h-咔唑的无水n,n-二甲基甲酰胺溶液缓慢滴加到含有叔丁醇钾的无水n,n-二甲基甲酰胺溶液中。
[0058]
在室温下搅拌2小时后,在15分钟内将含有2-溴-1,3-二氟苯的无水n,n-二甲基甲酰胺溶液滴入其中,配置成混合液a,混合液a中3,6-二叔丁基咔唑、叔丁醇钾、2-溴-1,3-二氟苯的摩尔比为2.2:2.7:1。
[0059]
将溶液加热到140℃搅拌24小时,然后冷却。将所得溶液倒入冰水中。混合物用二氯甲烷萃取,合并的有机层用盐水洗涤,经硫酸钠干燥,过滤并真空浓缩。
[0060]
使用石油醚的洗脱液通过柱色谱进一步纯化,得到中间产物m1为白色固体,重量9.1g,产率为80%。式(1)为中间产物m1的反应方程式。
[0061][0062]
s2.在氮气氛围保护下,将正丁基锂的正己烷溶液缓慢加入到含中间体m1的甲苯溶液中。
[0063]
缓慢升温到60℃,搅拌2小时后,真空下除去正己烷。
[0064]
降温到-40℃,加入三溴化硼,将反应混合物在室温下搅拌0.5小时。
[0065]
在0℃下加入n,n-二异丙基乙胺,得到混合液b,混合液b中正丁基锂、中间体m1、三溴化硼、n,n-二异丙基乙胺的摩尔比为2:1:2:2。
[0066]
将溶液加热到120℃搅拌24小时,冷却。
[0067]
将所得溶液倒入冰水中。混合物用二氯甲烷萃取,合并的有机层用盐水洗涤,经硫酸钠干燥,过滤并真空浓缩。然后使用石油醚洗脱液通过柱色谱进一步纯化,得到白色固体重量2.1g,产率为26%。式(2)为中间产物m2的反应方程式。
[0068][0069]
s3.将4,4'-二叔丁基-2,2'-二吡啶、1,5-环辛二烯氯化铱二聚体、中间体m2、联硼酸频那醇酯依次加入到超干四氢呋喃溶液中,得到混合液c,混合液c中4,4'-二叔丁基-2,2'-二吡啶、1,5-环辛二烯氯化铱二聚体、中间体m2和联硼酸频那醇酯的摩尔比为0.02:0.01:1:1.02。
[0070]
将混合液c用氮气鼓泡5分钟并加热到70℃,搅拌24小时,反应结束冷却至室温后。
[0071]
将反应混合物直接减压浓缩并通过柱层析纯化,使用二氯甲烷/石油醚(1:4)的混合物洗脱液,得到黄色固体重量4.50g,产率为90%。式(3)为中间产物m2的反应方程式。
[0072][0073]
s4.将4-溴-n,n-二苯基苯胺、中间体m3、碳酸钾加入到水和的四氢呋喃混合溶液中。
[0074]
将混合物再用氮气鼓泡5分钟,并在高氮气流下加入四三苯基膦钯,得到混合液d,混合液d内中间体m3、4-溴-n,n-二苯基苯胺、四三苯基膦钯和碳酸钾摩尔比为1:1.2:0.05:2。
[0075]
将混合物加热至80℃并搅拌12小时,反应结束冷却至室温。
[0076]
反应混合物用二氯甲烷和水萃取,合并的有机层真空浓缩,粗品进一步通过柱层析纯化,使用二氯甲烷/石油醚(1:10)得到黄色固体质量239.8mg,产率为55%。式(4)为中间体m3合成化合物a1的反应方程式。
[0077][0078]
实施例2
[0079]
一种具有窄谱带发射的芳基硼氮类化合物,化合物化学结构式如a2
[0080][0081]
具有窄谱带发射的芳基硼氮类化合物a2的制备方法,与实施例1不同之处在于,步骤s4中用3-溴-n,n-二苯基苯胺替代4-溴-n,n-二苯基苯胺。式(5)为中间体m3合成化合物a2的反应方程式。
[0082][0083]
性能测试
[0084]
(1)核磁共振氢谱
[0085]
采用布鲁克400mhz超导核磁共振仪分别对实施例1~2所得产物a1和a2进行核磁共振扫描,得到图1~2的1hmnr图。
[0086]
从图1可知,1h nmr(400mhz,cdcl3)δ=9.06(d,j=1.7,2h),8.44
–
8.35(m,6h),8.24(d,j=1.9,2h),7.79(d,j=8.6,2h),7.65(dd,j=8.8,2.0,2h),7.35(t,j=7.9,4h),7.31
–
7.25(m,5h),7.10(t,j=7.3,3h),1.64(d,j=8.0,18h),1.53(s,18h)。分子氢谱波峰能与实施例1目标产物a1一一对应,数量合理;
[0087]
从图2可知,1h nmr(400mhz,cdcl3)δ=9.10(d,j=1.7,2h),8.47
–
8.38(m,4h),8.27
–
8.19(m,4h),7.73(s,1h),7.61(dd,j=8.8,2.0,2h),7.54(s,1h),7.46(s,1h),7.38(t,j=7.8,4h),7.29(d,j=7.5,4h),7.23
–
7.19(m,1h),7.14(t,j=7.3,2h),1.66(d,j=6.6,18h),1.54(d,j=8.0,18h)。分子氢谱波峰能与实施例2目标产物a2一一对应,数量合理。
[0088]
(2)质谱
[0089]
将实施例1~2所得产物a1和a2放入液相质谱联用仪中,进行检测得到图3~4的质谱图。从图3可知,实施例1所得产物的相对分子质量为884.51,与所合成的一种具有窄谱带发射的芳基硼氮类化合物(a1)的相对分子质量一致;从图4可知,实施例2所得产物的相对分子质量为相对分子质量为884.54,与所合成的一种具有窄谱带发射的芳基硼氮类化合物(a2)的相对分子质量一致。
[0090]
(3)单晶x-射线衍射
[0091]
单晶x-射线衍射数据表明,化合物a1属于三斜晶系,空间群为p-1,z=4,
ɑ
=70.610(3)
°
,β=74.843(3)
°
,γ=72.578(3)
°
,v=5436.1(4);化合物a2属于正交晶系,空间群为p-1,z=2,
ɑ
=112.8090(10)
°
,β=97.4810(10)
°
,γ=104.9880(10)
°
,v=2401.89(5)。从图5所示化合物a1和a2的晶体的结构和作用力图中可以看到,所嵌入的硼中心都与核心骨架呈现出完美的三角平面共轭,促进了多重共振效应以及大的振子强度值。同时,单键连接的外围苯基表现出高度扭曲的几何形状,并在硼氮核心和外部部分之间产生大的空间位阻,二面角为26.58
°‑
31.01
°
,有利于实现较好的发光。从图6和图7可知,在分子的堆积中,化合物a1和a2存在着2种堆积模式,分别为头对尾和头对头的堆积,同时产生了较强c-h
…
π相互作用和弱的π-π相互作用,这些二聚体堆叠模式可以增强分子刚性并显着阻碍由分子内扭转引起的非辐射失活。
[0092]
因此,基于核磁共振、质谱和单晶x-射线衍射的结果,可以确定实施例1~2制备所得化合物的结构式分别如下a1、a2所示:
[0093][0094]
(4)紫外可见吸收光谱和荧光光谱
[0095]
紫外可见吸收光谱:将实施例1~2所得产物a1和a2分别溶于甲苯溶液中,配成1
×
10-3
mol/l的母液,采用岛津紫外可见光分光光度计uv-2700进行测试时,分别稀释成1
×
10-5
mol/l,测试得到a1和a2在甲苯溶液中的紫外可见吸收光谱图。参数设置;设置扫描范围是250~700nm。
[0096]
荧光光谱:采用爱丁堡fl980瞬态稳态荧光磷光光谱仪对实施例1~2所得产物分别进行测试得到a1和a2的荧光发射光谱图。参数设置:设置激发波长345nm,设置狭缝宽度,使其纵坐标数值接近一百万,进行光谱测试获得谱图。
[0097]
荧光光谱:采用爱丁堡fl980瞬态稳态荧光磷光光谱仪对实施例1~2所得产物分别进行测试得到a1和a2的荧光发射光谱图。参数设置:设置激发波长345nm,设置狭缝宽度,使其纵坐标数值接近一百万,进行光谱测试获得谱图。
[0098]
图8为实施例1所得产物a1在甲苯溶液中的紫外可见吸收光谱图和荧光发射光谱图,图9为实施例2所得产物a2在甲苯溶液中的紫外可见吸收光谱图和荧光发射光谱图。从图8~9可以看出,a1和a2的最大吸收峰位置分别均位于471nm和473nm,主要来自于短程的分子内电荷转移的跃迁,而400nm内的吸收峰,主要来自于骨架的跃迁。并且,a1的荧光发射峰位置为487nm,全半峰宽为22nm,斯托克斯位移为16nm,a2的荧光发射峰位置为489nm,全半峰宽为23nm,斯托克斯位移为16nm,说明化合物a1和a2具有典型的多重共振效应。此外,化合物a1和a2的斯托克斯位移非常小,表明其结构的刚性以及基态到激发态的结构弛豫小,有利于其辐射跃迁,实现较好的发光。
[0099]
(5)溶液荧光强度变化
[0100]
图10~11为25℃下,实施例1~2所得产物(a1、a2)在甲苯溶液中分别鼓入氮气和氧气后荧光强度变化的荧光光谱图。从图10~11可知,与在氮气气氛下相比,a1和a2在氮气气氛下表现出更高的荧光发射强度,并且发射的主峰没有发生变化,说明氧气会使所述一种具有窄谱带发射的芳基硼氮类化合物的a1和a2发生三重态的荧光猝灭,降低其荧光发射强度,表明所述一种具有窄谱带发射的芳基硼氮类化合物的a1和a2能够利用来自三重态的激子,实现更高的荧光量子产率。
[0101]
(6)单重态和三重态能级差的测试
[0102]
图12~13是实施例1所得产物在甲苯溶液中常温荧光和低温磷光图。从图12可以得到a1的单重态和三重态能级分别为2.53ev和2.42ev,进而得到了其单重态和三重态能级差为0.11ev。而从图13可以得到a2的单重态和三重态能级分别为2.54ev和2.50ev,进而得到了其单重态和三重态能级差为0.04ev。说明a2具有更小的单重态和三重态能级差,其更加容易实现热活化延迟荧光的性质。
[0103]
(7)热重分析
[0104]
采用高温同步热分析仪对一种具有窄谱带发射的芳基硼氮类化合物a1和a2分别进行热重分析,得到图14的热重分析图。测定条件:在氮气保护下,升温速率为10℃/min,测量温度范围为30~800℃。
[0105]
从图14可知,一种具有窄谱带发射的芳基硼氮类化合物a1和a2分别表现出高达490和493℃的热分解温度(td),说明其能在较高的温度下相对稳定,具有较好的热稳定性,为真空蒸镀工艺制作器件提供了必要的条件。
[0106]
制备实施例1
[0107]
有机光致发光材料a1的制备:采用真空蒸镀的方法,利用真空蒸镀手套箱将化合物a1和商用主体材料mcbp分别加热到升华温度,然后控制化合物a1和商用主体材料mcbp的升华速率比得到相应掺杂浓度的薄膜,最后将得到的薄膜进行退火处理即可。化合物a1占发光材料薄膜的质量占比为1%。
[0108]
薄膜的荧光光谱:利用爱丁堡fl980瞬态稳态荧光磷光光谱仪对薄膜进行测试,得到图15的荧光发射光谱。从图15中(a)图可知化合物a1在制备成薄膜后,其荧光发射峰位置为490nm,全半峰宽为26nm;说明化合物a1具有非常窄的全半峰宽,即化合物的色纯度高。发光材料的详细光致发光性能数据列于表3中。
[0109]
薄膜荧光寿命测试:采用爱丁堡fl980瞬态稳态荧光磷光光谱仪对实施例1所得产物分别进行测试。实验采用准分子激光器产生紫外光来激发样品,样品被激发出的荧光通
过望远镜系统进入光电倍增管,由光电倍增管引出的信号进入信号积分器,再进入计算机进行数据的采集和处理,测定条件为:激发脉冲重复频率为1000hz,脉宽为10ns,中心波长为375nm。
[0110]
荧光量子产率测试:爱丁堡fl980瞬态稳态荧光磷光光谱仪;测试方法:参数设置,用产物的最佳激发波长375nm激发,硫酸奎宁做参比,保持激发与发射的狭缝宽度一致,结果测得制备实施例1制得薄膜的荧光量子产率为95%,表明本发明的一种具有窄谱带发射的芳基硼氮类化合物具有较优的荧光量子产率。
[0111]
制备实施例2~6
[0112]
制备实施例2~6与制备实施例1基本相同,不同之处在于发光材料制备过程时掺杂化合物的种类和质量占比有所不同,具体各个制备实施例制备发光材料时掺杂化合物种类和质量占比见表1。
[0113]
表1制备实施例2~6制备发光材料时掺杂化合物种类和掺杂量占比
[0114] 掺杂化合物掺杂质量占比制备实施例2a21%制备实施例3a110%制备实施例4a120%制备实施例5a130%制备实施例6a230%
[0115]
制备对比例1~4
[0116]
制备对比例1~4与制备实施例1基本相同,不同之处在于发光材料制备过程时掺杂化合物的种类和质量占比有所不同,具体各个制备实施例制备发光材料时掺杂化合物种类和质量占比见表2。利用爱丁堡fls980瞬态稳态荧光磷光光谱仪对制备对比例1~4所得薄膜进行测试,得到图16的荧光发射光谱。
[0117]
表2制备对比例1~4制备发光材料时掺杂化合物种类和掺杂量占比
[0118] 掺杂化合物掺杂质量占比制备对比例1dtczb11%制备对比例2dtczb10%制备对比例3dtczb20%制备对比例4dtczb30%
[0119]
注:1,掺杂化合物dtczb结构式为:
[0120][0121]
表3:制备实施例1~6和制备对比例1~4所提供的光致发光材料的数据参数
[0122] 荧光发射峰位置/nm荧光量子产率%
制备实施例149095制备实施例249392制备实施例349494制备实施例449492制备实施例549488制备实施例649587制备对比例148892制备对比例249283制备对比例350160制备对比例450555
[0123]
表3中发光材料的详细光致发光性能数据表明,在制备实施例的化合物a1或a2和制备对比例的化合物dtczb掺杂比例都是1%时,所制备的发光材料的荧光量子产率高达95%和92%,制备对比例虽然在低浓度掺杂客体材料的情况下可以达到高的荧光量子产率,但是制备成器件时考虑到低浓度掺杂时所要求的真空蒸镀制备工艺条件要求更加苛刻,难以控制掺杂比例,其在制备成器件后的效率是比较低的。并且随着掺杂比例的升高,制备对比例的发光材料的荧光量子产率迅速下降,这是由于通过在分子骨架外围引入不同位置取代的基团三甲基苯,三甲基苯位阻小并且不能像化合物a1和a2所引入的三苯胺那样旋转,导致荧光淬灭的发生,无法与主体材料mcbp进行高浓度的掺杂,故制备对比例所述化合物在高浓度比例掺杂下其荧光量子产率迅速降低,导致其发光效率显著降低。而使用本技术提供的具有窄谱带发射的芳基硼氮类化合物作为客体掺杂材料时,将发光材料中化合物掺杂比例高达30%,制备实施例中发光材料的荧光量子产率维持在87%,掺杂比例控制容易,其在制备成器件的发光效率可以达到更高。
[0124]
图17为化合物a1和a2在掺杂薄膜的荧光寿命情况图。从图17可知,a1和a2同时具有瞬时荧光寿命和延迟荧光寿命。图18制备对比例1~4荧光寿命情况图。由于三重态激子从三重态回到单重态的过程中会发生激子的碰撞即三重态——三重态湮灭会导致激子的损失和严重效率滚降,并且延迟荧光的寿命越长,产生三重态——三重态湮灭的可能性越大。从图17和18可以看出,相比于对比例化合物,化合物a1和a2的延迟寿命都更加短,说明化合物a1和a2能够抑制三重态——三重态湮灭的产生,进而避免激子的损失和严重的效率滚降,实现更高的发光效率。
[0125]
更进一步地,通过图19制成薄膜的温度依赖的荧光寿命图可知,a1和a2的延迟荧光寿命随着温度的升高,延迟寿命的组分也逐渐的增加,a1和a2同时具有热活化延迟荧光的性质。表明所述具有窄谱带发射的芳基硼氮类化合物a1和a2能够利用来自三重态的激子,突破传统的荧光材料的25%的激子利用,达到91%以上,实现更高的荧光量子产率,从而使得其发光效率更高。
[0126]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1