具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂、制备方法及应用
1.本发明属于生物医药技术领域,涉及光敏剂,具体涉及一类具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂、制备方法及其在成像引导的光动力学治疗中的应用。
背景技术:
2.癌症已成为威胁社会进步和人类发展的主要健康问题。恶性细胞的快速增殖和扩散使癌症治疗异常困难。传统的癌症治疗方式,如手术切除、放疗和化疗等,都有很大的局限性,如侵袭性或不良副作用。光动力学治疗(pdt)作为一种无创、时空可控和非侵入的治疗手段,成为实现个性化精准治疗癌症的一个有效策略。其原理是光敏剂(pss)在特定光照射下产生活性氧物种(ros),其中以单线态氧(1o2)居多,它能破坏肿瘤细胞中的遗传物质,从而导致细胞凋亡或坏死。由此可见,光敏剂的性能对实现高效pdt起着关键作用。
3.在众多光敏剂中,具有光敏特性的小分子有机荧光团可用于肿瘤成像、肿瘤内部光敏剂的跟踪和可视化,因其更好的生物相容性、可降解性、易于调整的结构和光学性能而脱颖而出。而传统光敏剂由于平面刚性π共轭结构和较差的水溶性会导致聚集荧光猝灭效应(acq),从而降低了荧光发射强度和单线态氧(1o2)的生成效率,限制了其在生物医疗系统中的广泛应用。令人兴奋的是,2001年唐本忠院士团队首次提出聚集诱导发光(aie)概念,成功克服了acq荧光团的缺陷。更重要的是,聚集诱导发光体(aiegens)不仅有利于荧光成像,而且聚集态下,扭曲的三维结构可阻塞非辐射弛豫通道,可增加三重态量子产率和稳定性,进而提升ros的生成效率。
4.迄今为止,已经成功开发出多种聚集诱导发光型光敏剂(aie pss),并取得了可喜的进展,但是目前报道的aie pss发射波长大多还是在可见光范围内,这对实现高信噪比和深层组织穿透是十分不利的。在诊断技术中,近红外荧光成像具有可忽略生物底物自身荧光干扰、组织穿透能力强、直观、对比度高的显著优势。然而近红外aie pss的制备通常涉及多步反应、严苛的合成条件和费时的纯化或分离。除此之外,癌细胞的增殖与凋亡和线粒体的功能紧密相关。近年来,许多研究人员将研究重点放在了借助线粒体靶向功能提升光敏剂治疗功效上。研究结果表明,促进线粒体内1o2的生成可以有效地改善活性氧扩散距离短、寿命低的问题,从而显著提升治疗效果。尽管具有长波长发射的线粒体靶向的aie体系亦有不少报道,但其临床应用还受到了活性氧物种的产率(光敏性能)的限制。因此,为了将高性能aie pss进一步推向临床应用,开发新型的、具有线粒体靶向性能的近红外聚集诱导发光型超高效光敏剂意义重大。
技术实现要素:
5.本发明为了克服上述现有技术存在的缺陷进行,合成了一类具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂,并提供了该类光敏剂的制备方法,实现近红
外荧光成像引导的光动力学治疗。
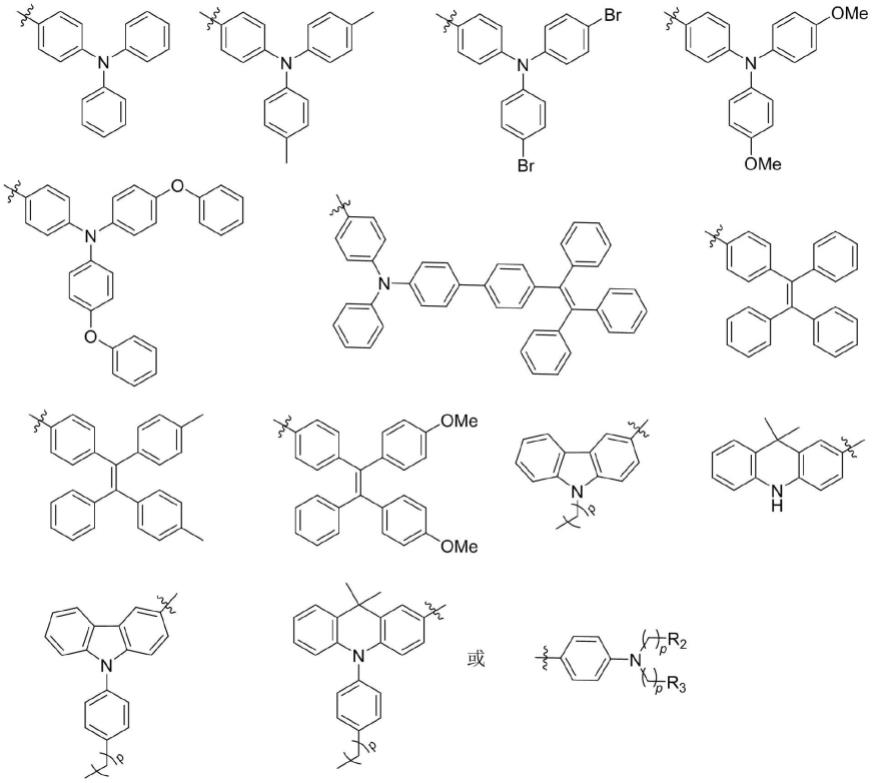
6.本发明的第一方面,提供一种具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂,该光敏剂是以不对称二乙腈基结构为核心设计的电子给体(d)-π-电子受体(a)型化合物,具有以下通式结构:
[0007][0008]
x,y包括c、n、o或s;
[0009]
m、n为0~3的自然数,且不同时为0;
[0010]
ar1选自下述任一结构:
[0011][0012]
其中,p为1~5的自然数,r2和r3分别各自独立地选自h、羟基、胺基、任选被取代的烷基、烷氧基、烷硫基、烯基、炔基、环烷基、环烷基氧基、环烷基硫基、酰胺基、芳基、杂环基、杂芳基、杂环烷基、单烷基胺基或二烷基胺基中的一个;
[0013]
ar2选自下述任一结构:
[0014][0015]
z独立地选自卤素根、醋酸根、三氟乙酸根、磷酸二氢根、硫酸氢根、四氟硼酸根、六氟磷酸根、磺酸根中的一个;
[0016]
r1为直链烷基、环烷基、羧基取代烷基、醛基取代烷基、磺酸基取代烷基、叠氮取代烷基、季胺取代烷基,优选ch3、ch2ch3、ch2cooh、ch2cho、ch2ch3so3或ch2ch3n3;r2和r3分别独立地选自h、nh3、oh、och3、och2ch3或conh2中的一种。
[0017]
本发明的第二方面,提供上述光敏剂的制备方法,制备路线如下:
[0018][0019]
制备方法包括如下步骤:
[0020]
s1、将对芳基二乙腈(ⅱ)与芳基甲醛(ⅲ)进行knoevenagel缩合反应,得到所述中间产物ⅳ;
[0021]
s2、将中间产物(ⅳ)与n取代芳基甲醛(
ⅴ
)再进行一步knoevenagel缩合反应,得到不对称二乙腈基中间产物(ⅵ);
[0022]
s3、将中间产物(ⅵ)再与卤代烷烃、磺酸内酯、磺酸基取代烷烃或其他各种取代烷烃发生取代或成盐反应,得到ⅰ所示结构式中的目标产物。
[0023]
各制备步骤的优选条件如下:
[0024]
s1步骤中的knoevenagel缩合反应具体过程为:在惰性气体保护下,将对芳基二乙腈(ⅱ)与芳基甲醛(ⅲ)加入有机溶剂中,再添加少量碱性物质作催化剂,之后室温反应6~12h。
[0025]
优选地,ⅱ与ⅲ的物质的量之比为1:1~4;碱性物质的摩尔量为0.1~0.5当量,选自吡啶、哌啶、碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、醇钠、醇钾中的任一种或多种混合物;惰性气体为氮气;所用有机溶剂选自乙醇、异丙醇、四氢呋喃、乙腈中的一种或几种。
[0026]
s2步骤中的knoevenagel缩合反应具体过程为:在惰性气体保护下,将中间产物(ⅳ)与n取代芳基甲醛(
ⅴ
)加入有机溶剂中,再添加少量碱性物质作催化剂,之后升温到回流反应温度,反应6~12h。
[0027]
优选地,中间产物(ⅳ)与芳基甲醛(
ⅴ
)的摩尔比为1:1~2,碱性催化剂比例及用量、有机溶剂和惰性气体同s1步骤,回流反应温度为70~120℃,反应时间为6~12h。
[0028]
产物分离、提纯方法具体如下:得到的混合物冷却到室温,经由减压浓缩所得到的粗产品,再加入热的有机溶剂(如乙醇、甲醇)洗涤多次,过滤得中间产物ⅵ。
[0029]
s3成盐反应过程中,取代或成盐反应在惰性气体保护下进行,反应物溶于有机溶剂中,接着升温至回流反应温度,反应12~24h。
[0030]
优选的,中间产物ⅵ与卤代烷烃、磺酸内酯、磺酸基取代烷烃或其他各种取代烷烃的摩尔比为1:1~5;回流反应的温度为90~150℃,惰性气体为氮气;采用的有机溶剂为1,4-二氧六环、乙腈、n,n
′‑
二甲基甲酰胺、二甲亚砜、丙酮或甲苯中的一种或几种。
[0031]
该步骤产物分离、提纯方法具体如下:反应得到的混合物冷却到室温,经由减压浓缩所得到的粗产品,再加入乙酸乙酯进行少量多次洗涤,过滤得到目标化合物。
[0032]
本发明提供具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂,具有聚集诱导发光性能,且最大发射波长位于近红外区,可用于近红外荧光成像,并且能够特异性靶向线粒体,是一种优异的线粒体成像对比剂。
[0033]
此外,该光敏剂在光源辐照下,能在细胞内超高效地产生单线态氧,有效地杀死肿瘤细胞。
[0034]
因此,本发明的第三方面,提供了该光敏剂在制备光动力学治疗剂中的用途,尤其在近红外荧光成像引导的光动力学治疗剂方面的应用。
[0035]
从光敏剂结构来说,本发明以对芳基二乙腈为结构基元,通过改变分子结构ar1位的给电子基团(a)和ar2位的吸电子基团(d)并辅以r1位的柔性离子团合成一系列具有聚集诱导发光性能的超高效光敏剂。吸电子基(a)和给电子基团(d)通过共轭的π体系进行桥连,能够增强d-π-a效应,这不仅使探针拥有近红外发光,另一方面化合物的homo(最高占有分子轨道)和lumo(最低未占有分子轨道)的电子云分布会显著分离,从而有利于1o2的产生,使得以该化合物作为光敏剂就能实现近红外成像引导的高效光动力学治疗。另外,柔性阳离子链的引入不仅使该类光敏剂具备了特异性靶向线粒体的能力,还增强了光敏剂的生物相容性,更有利于生物应用。从探针性能来说,该类化合物显示出近红外aie特性,具有穿透性强、背景干扰低、光损伤小等优点,在光动力学治疗领域表现出了很大的潜力。
[0036]
与现有技术相比,本发明提供的具有不对称二乙腈基结构的光敏剂实现了以下有益效果:(1)该光敏剂的合成方法简单、原料来源方便易得且具有较高的产率;(2)该光敏剂具有aie特性,最大吸收峰在可见光区,普通白光就能激活该类aie光敏剂;(3)该光敏剂具有近红外发射(>650nm),用于荧光成像时,近红外荧光信号穿透性能强、有利于消除成像时生物组织自体荧光的干扰;(4)该光敏剂具有超高的单线态氧产率,可达玫瑰红b(一种常见的测定单线态氧产率的参比化合物)的6倍以上;(5)本发明提供的具有近红外aie性能的光敏剂能特异性地标记活细胞中的线粒体,线粒体中产生的1o2能够短时间内杀死癌细胞,提高了pdt效率;(6)本发明所述的光敏剂还可以实现动物水平的活体成像,通过瘤内注射入体内后,可逐渐在肿瘤部位富集,呈现良好的荧光成像能力;(7)该光敏剂具有较好的光稳定性和生物相容性,较小的暗毒性,通过构建荷瘤鼠之后,可以用于荷瘤鼠近红外荧光成像引导的光动力治疗。
附图说明
[0037]
图1为实施例1制备的化合物tpa-py-pf6的核磁共振氢谱表征结果;
[0038]
图2为实施例1制备的化合物tpa-py-pf6的核磁共振碳谱表征结果;
[0039]
图3为实施例1制备的化合物tpa-py-pf6的高分辨质谱表征结果;
[0040]
图4为实施例2制备的化合物tpe-py-pf6的核磁共振氢谱表征结果;
[0041]
图5为实施例2制备的化合物tpe-py-pf6的核磁共振碳谱表征结果;
[0042]
图6为实施例2制备的化合物tpe-py-pf6的高分辨质谱表征结果;
[0043]
图7为实施例3制备的化合物dea-py-pf6的核磁共振氢谱表征结果;
[0044]
图8为实施例3制备的化合物dea-py-pf6的核磁共振碳谱表征结果;
[0045]
图9为实施例3制备的化合物dea-py-pf6的高分辨质谱表征结果;
[0046]
图10为实施例1、2、3制备的tpa-py-pf6、tpe-py-pf6和dea-py-pf6在二甲基亚砜(浓度为10μm)中的紫外-可见吸收光谱图;
[0047]
图11为实施例1、2、3制备的tpa-py-pf6、tpe-py-pf6和dea-py-pf6在二甲基亚砜与甲苯的混合溶剂(体积比为1:9)中的荧光发射光谱图;
[0048]
图12为实施例1制备的tpa-py-pf6在dmso与甲苯混合体系中的荧光发射光谱图与相对荧光强度图;
[0049]
图13为实施例2制备的tpe-py-pf6在dmso与甲苯混合体系中的荧光发射光谱图与相对荧光强度图;
[0050]
图14为实施例3制备的dea-py-pf6在dmso与甲苯混合体系中的荧光发射光谱图与相对荧光强度图;
[0051]
图15为实施例1制备的tpa-py-pf6的单线态氧检测图;
[0052]
图16为实施例2制备的tpe-py-pf6的单线态氧检测图;
[0053]
图17为实施例3制备的dea-py-pf6的单线态氧检测图;
[0054]
图18为实施例7制备的tpa-py-so3的单线态氧检测图;
[0055]
图19为不同光敏剂存在的情况下,abda的吸收衰减曲线图,其中a0为初始吸光度,a为光照后的最终吸光度;
[0056]
图20为本发明实施例10中,tpa-py-pf6在小鼠4t1细胞内产生1o2的性能测试图,(a)和(b)分别为荧光场和明场的激光共聚焦图像;
[0057]
图21为本发明实施例10中,tpe-py-pf6在小鼠4t1细胞内产生1o2的性能测试图,(a)和(b)分别为荧光场和明场的激光共聚焦图像;
[0058]
图22为本发明实施例10中,dea-py-pf6在小鼠4t1细胞内产生1o2的性能测试图,(a)和(b)分别为荧光场和明场的激光共聚焦图像;
[0059]
图23为本发明实施例11中,tpa-py-pf6在小鼠4t1细胞内的线粒体共定位测试图;其中,(a)为tpa-py-pf6的荧光信号;(b)为商业线粒体染料的荧光信号;(c)为(a)和(b)的混合场;(d)为(a)和(b)的重叠系数;
[0060]
图24为本发明实施例11中,tpe-py-pf6在小鼠4t1细胞内的线粒体共定位测试图;其中,(a)为tpe-py-pf6的荧光信号;(b)为商业线粒体染料的荧光信号;(c)为(a)和(b)的混合场;(d)为(a)和(b)的重叠系数;
[0061]
图25为本发明实施例11中,dea-py-pf6在小鼠4t1细胞内的线粒体共定位测试图;
其中,(a)为dea-py-pf6的荧光信号;(b)为商业线粒体染料的荧光信号;(c)为(a)和(b)的混合场;(d)为(a)和(b)的重叠系数;
[0062]
图26为本发明实施例12中,在光照(white light)或避光(dark)下,与不同浓度(0、1、2、5、10、20、50、100μmol/l)的tpa-py-pf6共存时,4t1细胞的存活率;
[0063]
图27为本发明实施例12中,在光照(white light)或避光(dark)下,与不同浓度(0、1、2、5、10、20、50、100μmol/l)的tpe-py-pf6共存时,4t1细胞的存活率;
[0064]
图28为本发明实施例12中,在光照(white light)或避光(dark)下,与不同浓度(0、1、2、5、10、20、50、100μmol/l)的dea-py-pf6共存时,4t1细胞的存活率;
[0065]
图29为本发明实施例13中,tpa-py-pf6在4t1荷瘤小鼠的肿瘤部位的近红外荧光成像性能测试图,(a)图为实施例1制得的光敏剂tpa-py-pf6瘤内注射到4t1荷瘤小鼠体内后,对于不同处理组,小鼠体内tpa-py-pf6的荧光信号随时间的变化图;(b)图为pbs或光敏剂处理4t1荷瘤小鼠24h后,将4t1荷瘤小鼠解剖获得离体组织(肿瘤、心、肝、脾、肺、肾)的近红外荧光成像对比图;
[0066]
图30为本发明实施例14中,不同处理组对4t1荷瘤小鼠光动力肿瘤治疗效果对比曲线图,(a)小鼠肿瘤体积和(b)小鼠体重随天数变化的曲线;
[0067]
图31为本发明实施例14中各组治疗12天后的肿瘤大小对比图。
具体实施方式
[0068]
本发明的目的、优点和特点,将通过下面优选实施例的非限定性说明进行图示和解释,此处结合附图所举实例仅用以解释本发明,并非用于限定本发明的实施范围。下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
[0069]
对于在化学、生物化学或相关领域的技术人员来说,在不脱离本发明宗旨和权利要求所保护的范围情况下,还可做出很多修改和变换,这些均属于本发明的保护范围之内。
[0070]
声明:本发明所使用的原材料均为普通市售。
[0071]
本发明采用以下合成路线制得具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂:
[0072]
[0073]
第一步,在氮气保护下,将对芳基二乙腈(ⅱ)与芳基甲醛(ⅲ)按照1:(1~4)的摩尔比溶于有机溶剂中,再加入碱性物质作为催化剂,室温反应6~12h,得到中间产物(ⅳ)。
[0074]
第二步,在氮气保护下,将第一步反应得到的中间产物(ⅳ)和n取代芳基甲醛(
ⅴ
)溶于有机溶剂,ⅳ与
ⅴ
的摩尔比为1:(1~2),再加入碱性物质作为催化剂,于70~120℃回流反应,得到ⅵ所示的结构式的中间产物。
[0075]
第三步,在氮气保护下,将第二步反应的中间产物(ⅵ)与卤代烷烃、磺酸内酯、磺酸基取代烷烃或其他各种取代烷烃按照1:(1~5)的摩尔比溶于有机溶剂,90~150℃回流反应,得到目标化合物。
[0076]
实施例1
[0077]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂tpa-py-pf6(i-1)的制备方法,步骤如下:
[0078]
(1)将468.2mg对苯二乙腈(ⅱ,3.0mmol)添加到三口烧瓶中,加入7.5mlthf和2.5ml etoh。将237.1mg 4-(二苯胺)苯甲醛(
ⅲ‑
1,1.0mmol)溶于15mlthf中,20mg氢氧化钠溶于5ml etoh中,二者混合均匀后加入恒压滴液漏斗中。抽真空充氮气三次之后,缓慢打开恒压滴液漏斗,室温搅拌反应12h。反应液加入50ml二氯甲烷中萃取三次,合并有机相后用饱和食盐水洗涤三次,无水硫酸钠干燥。减压蒸馏除掉有机溶剂,粗产物用硅胶柱色谱分离纯化,洗脱液为石油醚:乙酸乙酯=10:1(体积比),得到255mg黄绿色固体为(z-2-(4-(氰甲基)苯基)3-(4-(二苯胺)苯基)丙烯腈(
ⅳ‑
1),产率:51%。
[0079]
本步骤的反应式如下:
[0080][0081]
(2)将411.2mg
ⅳ‑
1、128.4mg 4-吡啶苯甲醛(
ⅴ‑
1,1.2mmol)和42.6μl哌啶加入40ml乙醇中,在氮气保护下,升温至回流反应12h。反应液冷却至室温后加入50ml二氯甲烷中萃取三次,合并有机相后用饱和食盐水洗涤三次,无水硫酸钠干燥。减压蒸馏除掉有机溶剂,粗产物用硅胶柱色谱分离纯化,洗脱液为石油醚:乙酸乙酯=1:1(体积比),得到480.3mg橙黄色固体为(z)-2-(4-((z)1-氰基-2-(4-(二苯胺)苯基)乙烯基)苯基)-3-(吡啶-4-丙烯腈)(
ⅵ‑
1),产率:85%。
[0082]
本步骤的反应式如下:
[0083][0084]
(3)将250.2mg
ⅵ‑
1(0.5mmol)和58μl碘乙烷(0.6mmol)加入15ml乙腈溶剂中,所得混合物在氮气保护下,升温至回流温度后再反应6h。反应液冷却至室温后减压蒸馏除掉有机溶剂,粗产物用15ml丙酮溶解后,加入5ml kpf6饱和水溶液,混合物在氮气保护下,升温至回流温度后反应3h。冷却至室温后,用dcm和水萃取三次,收集有机相,旋干溶剂后用乙酸
乙酯洗涤混合残渣,过滤得到的631.8mg黑色固体为tpa-py-pf6(i-1),产率:91%。
[0085]
化合物i-1的核磁共振谱表征和高分辨质谱表征参见图1~图3,数据如下:1h nmr(400mhz,dmso-d6)δ[ppm]:δ9.20(d,j=6.8hz,2h),8.48(d,j=6.7hz,2h),8.41(s,1h),8.10(s,1h),7.95(dt,j=16.7,8.9hz,6h),7.42(t,j=7.8hz,4h),7.20(dd,j=16.3,7.9hz,6h),6.97(d,j=8.9hz,2h),4.65(q,j=7.3hz,2h),1.59(t,j=7.3hz,3h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ149.97,145.80,145.26,135.20,131.22,129.93,128.39,127.49,127.11,126.19,125.82,124.96,119.48,83.89,24.61.hrms for c
37h29n4+
[m
–
pf6]
+
,calculated:529.2392;found:529.2392。
[0086]
本步骤的反应式如下:
[0087][0088]
实施例2
[0089]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂tpe-py-pf6(i-2)的制备方法,重复实施例1,区别在于将上述第(1)步中的4-(二苯胺)苯甲醛(
ⅲ‑
1)改为4-(1,2,2-三苯乙烯)苯甲醛(
ⅲ‑
2),其他条件不变。最终得到目标化合物tpe-py-pf6(i-2)的红色固体,产率:83%。
[0090]
化合物i-2的结构表征图谱参见图4~图6,数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ9.22(d,j=6.8hz,2h),8.48(d,j=6.7hz,2h),8.44(s,1h),8.12(s,1h),7.99(q,j=8.7hz,4h),7.78(d,j=8.4hz,2h),7.17(dt,j1=8.8hz,j2=4.2hz,11h),7.07
–
6.95(m,6h),4.66(q,j=7.3hz,2h),1.59(t,j=7.3hz,3h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ193.41,151.09,150.47,149.58,141.29,140.76,136.61,131.92,126.77,125.44,122.72,122.67,122.64,122.13,119.19,114.02,100.15,43.83,12.41.hrms for c
45h34n3+
[m
–
pf6]
+
,calculated:616.2753;found:616.2757。
[0091]
其合成路线如下:
[0092][0093]
实施例3
[0094]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂dea-py-pf6(i-3)的制备方法,重复实施例1,区别在于将上述第(1)步中的4-(二苯胺)苯甲醛(
ⅲ‑
1)改为4-(二乙氨基)苯甲醛(
ⅲ‑
3),其他条件不变。最终得到目标化合物dea-py-pf6(i-3)的黑色固体,产率:90%。
[0095]
化合物i-3的结构表征图谱参见图7~图9,数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ9.20(d,j=6.8hz,2h),8.47(d,j=6.8hz,2h),8.38(s,1h),8.10
–
7.80(m,7h),6.82(d,j=9.1hz,2h),4.65(q,j=7.3hz,2h),3.46(q,j=6.9hz,4h),1.58(t,j=7.3hz,3h),1.24
–
1.05(m,6h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ149.75,148.83,144.89,137.83,132.11,131.05,127.37,126.66,125.55,119.83,119.13,118.81,116.08,111.08,99.80,56.23,43.86,16.06,12.47.hrms for c
29h29n4+
[m
–
pf6]
+
,calculated:433.2392;found:433.2384。
[0096]
其合成路线如下:
[0097]
[0098]
实施例4
[0099]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂tpa-qu-pf6(i-4)的制备方法,重复实施例1,区别在于实施例1第(2)步中的4-吡啶苯甲醛(
ⅴ‑
1)改为4-喹啉苯甲醛(
ⅴ‑
2),最终得到目标化合物tpa-qu-pf6(i-4)。其合成路线如下:
[0100][0101]
化合物i-4的结构表征数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ9.01(s,1h),8.60(s,1h),8.42(s,1h),8.23(s,1h),8.10(s,1h),7.94(s,2h),7.89(s,2h),7.75(s,1h),7.33(s,2h),7.25(d,j=10.0hz,6h),7.18(s,2h),7.08(s,4h),7.00(s,2h),4.80(s,2h),1.57(s,3h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ146.93,145.86,143.71,143.27,143.14,142.60,142.43,134.49,132.57,131.93,130.39,129.80,129.27,128.36,126.82,124.67,124.23,124.18,123.15,122.99,121.32,121.16,112.55,108.47,54.30,12.57.hrms for c
41h31n4+
[m
–
pf6]
+
,calculated:579.2543;found:433.2534。
[0102]
实施例5
[0103]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂meo-tpa-py-pf6(i-5)的制备方法,步骤如下:
[0104]
(1)将500mg苯胺(vii,5.5mmol)、5.0g 1-碘-4-甲氧基苯(viii,22.0mmol)、1.3g铜(22.0mmol)、300mg 18-冠醚-6(1.1mmol)和6.0g k2co3(44.0mmol)在氮气下保护下置于完全干燥的两口烧瓶中。之后将50ml脱气的邻二氯苯注入烧瓶中,搅拌混合物并升温至回流温度反应48h。反应液冷却至室温后过滤,用二氯甲烷萃取三次,合并有机相后用饱和食盐水洗涤三次,无水硫酸钠干燥,减压蒸馏除掉有机溶剂。粗产物用硅胶柱色谱分离纯化,洗脱液为石油醚:乙酸乙酯=3:1(体积比),得到白色固体4,4
′‑
二甲氧基三苯胺(ix)为1.3g,产率:81%。
[0105]
本步骤的反应式如下:
[0106][0107]
(2)在0℃下,将1.0g 4,4
′‑
二甲氧基三苯胺(ix,3.3mmol)溶于20mldmf中,400μc39h33
n4o
2+
[m
–
pf6]
+
,calculated:589.2598;found:589.2566。
[0113]
实施例6
[0114]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂meo-tpa-qu-pf6(i-6)的制备方法,重复实施例5的前三步,合成(z)3-(4-(二(4-甲氧基苯基)氨基)苯)2-(4-(氰甲基)苯基)丙烯氰(
ⅳ‑
4)。之后重复实施例4的最后两步,区别在于将
ⅳ‑
1换成
ⅳ‑
4,其合成路线如下:
[0115][0116]
化合物i-6的结构表征数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ9.01(s,1h),8.60(s,1h),8.42(s,1h),8.23(s,1h),8.10(s,1h),7.92(d,j=25.0hz,4h),7.75(s,1h),7.33(s,2h),7.26(s,2h),7.18(s,6h),6.79(s,4h),4.80(s,2h),3.79(s,6h),1.57(s,3h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ160.03,145.86,143.71,143.27,143.14,142.60,142.43,138.07,134.49,132.57,131.93,130.39,129.80,128.36,127.01,126.82,124.23,124.18,123.15,121.32,121.16,114.48,112.55,108.47,56.08,54.30,12.57.hrms for c
43h35
n4o
2+
[m
–
pf6]
+
,calculated:639.2755;found:639.2759。
[0117]
实施例7:
[0118]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂tpa-py-so3(化合物i-7)的制备方法,区别在于实施例1第(3)步中的碘乙烷改为1,3-丙烷磺酸内酯,最终得到目标化合物tpa-py-so3(i-7)。本步骤的合成路线如下:
[0119][0120]
化合物i-7的结构表征数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ8.81(s,2h),7.92(d,j=25.0hz,4h),7.75(s,2h),7.33(s,2h),7.25(d,j=10.0hz,5h),7.18(s,3h),7.08(s,4h),7.00(s,2h),3.53(s,2h),2.57(d,j=20.0hz,4h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ146.93,145.86,145.24,142.60,140.47,134.49,131.93,130.39,129.27,126.54,124.67,124.23,123.15,122.99,121.16,108.47,59.27,47.83,23.84.hrms for c
38h30
n4o3s
+
[m+na]
+
,calculated:645.1936;found:645.1930。
[0121]
实施例8:
[0122]
具有不对称二乙腈基结构的近红外聚集诱导发光型超高效光敏剂meo-tpa-bt-pf6(i-8)的制备方法,区别在于实施例5第(2)步中的4-吡啶苯甲醛(
ⅴ‑
1)改为6-苯并噻唑甲醛(
ⅴ‑
3),最终得到目标化合物meo-tpa-bt-pf6(i-8),其合成路线如下:
[0123][0124]
化合物i-8的结构表征数据为:1h nmr(400mhz,dmso-d6)δ[ppm]:δ8.10(s,1h),7.89(s,2h),7.75(s,2h),7.46(s,1h),7.38(s,5h),7.18(s,6h),6.79(s,4h),5.89(s,1h),4.80(s,2h),3.79(s,6h),1.57(s,3h).
13
c nmr(100mhz,dmso-d6)δ[ppm]:δ163.90,160.03,145.86,143.21,142.60,138.07,135.95,135.17,134.49,134.46,131.93,130.39,127.01,126.96,124.23,124.01,123.15,121.16,119.33,114.48,110.88,108.47,56.08,53.20,13.62.hrms for c
41h33
n4o2s
+
[m
–
pf6]
+
,calculated:645.2319;found:645.2324。
[0125]
实施例9:光物理性能以及生成单线态氧的特性研究
[0126]
(1)光物理特性研究
[0127]
以实施例1、2和3中生成的光敏剂(tpa-py-pf6、tpe-py-pf6、dea-py-pf6)作为实施例进行说明,其它产品的研究同此实施例,不再赘述。
[0128]
将合成的光敏剂分别溶解在dmso溶液中,配制成浓度为10
–3m的母液,再将母液稀释成10
–5m的溶液以测试紫外吸收光谱和不同溶剂体积比的dmso/甲苯混合溶剂中荧光发射光谱的变化。如图10中的紫外-可见吸收光谱所示,tpa-py-pf6、tpe-py-pf6和dea-py-pf6的吸收波段很宽,最大吸收峰分别位于476nm、419nm和495nm处。如图11所示,tpa-py-pf6、tpe-py-pf6和dea-py-pf6的发射光谱分布在500
–
800nm处,最大发射波长位于800nm左右,属于近红外发光范畴。在dmso中荧光发射峰较弱,当逐步加入甲苯后,随着甲苯比例的增加,探针逐渐形成聚集体,使得分子内运动受到限制,有效地阻挡非辐射通道并激活其辐射跃迁,光敏剂的荧光强度明显增强。如图12
–
14所示,当甲苯含量达到90%时,光敏剂的最大发射波长分别位于660nm、783nm和768nm处。研究结果表示,三个荧光分子皆展现了典型的aie特性。另外,生成的荧光分子具有超过200nm的斯托克斯位移,避免了生物医学成像过程中激发光的干扰和发射光的自吸收。
[0129]
(2)生成单线态氧的能力
[0130]
以实施例1、2、3和7中生成的光敏剂(tpa-py-pf6、tpe-py-pf6、dea-py-pf6、tpa-py-so3)作为实施例进行说明,其它产品不再赘述。
[0131]
为了评估生成的光敏剂在光照下生成单线态氧(1o2)的能力,以商用的9,10-蒽二基-双(亚甲基)二丙二酸(abda)为指示剂,同时,选用广泛使用的光敏剂玫瑰红b(rb)作为标准参照,检测1o2的产生效率。如图15
–
18所示,当光敏剂存在时,用25mw/cm2的白光照射2分钟,abda在378nm处的紫外吸收呈现出快速下降的趋势,abda的吸光度下降速率越快,可以说明该光敏剂生成1o2速率较快。如图19所示,从abda的衰减曲线图可以明显地观察到,tpa-py-pf6和tpe-py-pf6光敏剂生成1o2的速率远高于商用光敏剂玫瑰红b。经计算,每分钟10.00nmol的tpa-py-pf6和tpe-py-pf6分别可降解abda的量为38.70nmol和32.60nmol,而相同条件下玫瑰红b可降解abda的量仅为8.75nmol。进一步证实了光敏剂tpa-py-pf6和tpe-py-pf6可以高效地产生活性氧。这意味着,在光动力治疗技术方面,该类aie荧光团作为光敏剂将具有良好的治疗潜力。
[0132]
实施例10:光敏剂在小鼠乳腺癌细胞内产生1o2能力的评价
[0133]
以绿色单线态氧传感器(sosg)作为活性氧指示剂,将小鼠乳腺癌细胞(4t1细胞)接种于共聚焦培养皿中,置于温度为37℃、体积浓度为5%的co2培养箱中培养过夜。之后旧培养基用含有实施例1、2或3中制得的tpa-py-pf6、tpe-py-pf6或dea-py-pf6的新鲜培养基溶液替换,继续培养1h。取出含药物的培养皿用白光(100mw/cm2)辐照不同时间(0、30s、1min、2min、5min、10min、15min或30min),接着更换含有sosg的新鲜培养基,培养1h。最后用pbs洗涤三次,加入1ml新鲜培养基,用激光共聚焦显微镜观察每个培养皿中4t1细胞内的绿色荧光强度。
[0134]
结果如图20a-22a所示,随着光照时间的延长,细胞在绿色通道的荧光强度逐渐增强。此外,如图20b-22b的明场图像显示,在白光照射15min后,用tpa-py-pf6或tpe-py-pf6处理的两组细胞几乎都变成圆形,胞质渗漏,细胞骨架破坏,并开始从培养板脱落。dea-py-pf6处理的细胞在光照20min后也出现类似的现象。综合以上结果证实了按照上述技术路线合成的光敏剂可以在光照条件下产生1o2,有效杀死肿瘤细胞的观点。
[0135]
实施例11:线粒体共定位能力评估
[0136]
将小鼠乳腺癌细胞(4t1细胞)接种于共聚焦培养皿中,置于温度为37℃、体积浓度为5%的co2培养箱中培养过夜。然后用商用线粒体生物探针(mitotracker deep red)与tpa-py-pf6、tpe-py-pf6或dea-py-pf6处理的两组细胞共染,确定光敏剂在癌细胞中的富集位置。利用共聚焦激光显微镜观察两种荧光信号的重叠程度。
[0137]
根据图23-25的结果显示,来自光敏剂的红色荧光信号与来自线粒体染料的绿色信号(假色)融合良好。表明它们具有靶向和成像癌细胞线粒体的能力,这有利于提高pdt效率。
[0138]
实施例12:tpa-py-pf6、tpe-py-pf6和dea-py-pf6对4t1细胞的杀伤力评价
[0139]
将处于对数生长期的细胞用胰酶消化后,用完全培养基稀释成细胞悬液,随之将其以1
×
104个/孔的密度接种于96孔板,置于37℃,5%的co2培养箱培养,12h后加入不同浓度的tpa-py-pf6、tpe-py-pf6或dea-py-pf6,使得样品终浓度分别为0、1、2、5、10、15、20、50和100μm,培养1h,然后白光光照(100mw/cm2)30分钟,与此同时,处于相同实验条件下不进行光照的实验组也进行暗毒性研究。再分别培养4h,12h和24h后,移走含光敏剂的培养基,接着用新鲜的含10%cck-8的培养基(无fbs)在黑暗条件下培养1h,然后用酶标仪测试450nm处的吸光度值(od值),相应的细胞存活率通过以下公式计算:细胞存活率(%)=
(od
样品
–
od
背景
)/(od
对照
–
od
背景
)
×
100%。
[0140]
不同浓度的光敏剂对4ti细胞的杀伤性如图26-28所示。在光照条件下,tpa-py-pf6、tpe-py-pf6和dea-py-pf6对4ti细胞的杀伤性具有浓度依赖性。当光敏剂的浓度低至10μmol/l时,细胞存活率达到20%以下,表明制备的光敏剂对4ti细胞显示出高效的光动力治疗作用。
[0141]
实施例13:肿瘤成像测试
[0142]
将pbs和实施例1制备得到的tpa-py-pf6(0.4mg/ml,50μl)通过瘤内注射到4t1荷瘤小鼠体内,利用小动物活体成像系统在注射后不同时间点采集小鼠照片。
[0143]
小鼠体内tpa-py-pf6的近红外荧光成像随时间的变化如图29a所示,可知,tpa-py-pf6的荧光强度在9h后趋于稳定,直至24h荧光强度没有明显减弱,体现了在肿瘤部位具有良好的药物保留和富集能力;注射tpa-py-pf624 h后,将4t1荷瘤小鼠解剖获得的离体组织(肿瘤和心、肝、脾、肺、肾)进行近红外荧光成像对比。如图29b所示,只有肿瘤部位有荧光信号,进一步说明tpa-py-pf6在肿瘤部位的大量富集。上述结果表明tpa-py-pf6可以在小鼠体内实现近红外荧光成像。
[0144]
实施例14:体内抗肿瘤评估
[0145]
将移植瘤4t1肿瘤小鼠随机分为4组,每组5只,包括pbs组、pbs+光照组、tpa-py-pf6组和tpa-py-pf6+光照组,通过瘤内注射打入100μltpa-py-pf6(8mg/kg)或生理盐水溶液。之后,光照组用白光照射20分钟进行治疗。在各种处理后,每天记录小鼠的肿瘤大小和体重。用游标卡尺测量肿瘤体积,按照公式v=(肿瘤长度
×
肿瘤宽度2)/2计算。为进一步评价光治疗效果,治疗12天后处死小鼠,取出不同治疗组的肿瘤组织,用游标卡尺测量肿瘤体积并称重。
[0146]
4组4t1荷瘤小鼠的肿瘤的光动力治疗结果如图30a所示,只有治疗组(tpa-py-pf6+光照组)具有抑制肿瘤生长甚至肿瘤消融的作用,如图30b所示,不同处理组小鼠的体重没有明显变化,说明tpa-py-pf6具有较低的暗毒性。处理12天后处死小鼠,收集并测量肿瘤体积。如图31所示,发现治疗组的肿瘤体积明显减少,存在完全治愈的情况。证明tpa-py-pf6具有在复杂的生物系统中有效地进行pdt的能力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1