一种可细胞内自组装的蛋白降解剂及其制备方法和应用
1.本发明属于药物制备技术领域,涉及一种可细胞内自组装的蛋白降解剂及其制备方法和应用。
背景技术:
2.linifanib是一种结构新颖的受体酪氨酸激酶(rtk)抑制剂,是血管内皮生长因子(vegf)和血小板衍生生长因子(pdgf)受体家族成员的有效抑制剂,抑制kdr,flt-1,pdgfrβ和flt3的ic
50
值分别为3,4,66,4nm。在体内实验中对于vegf和pdgf受体家族的成员,linifanib显示ic
50
值为4nm(kdr)至190nm(flt4),但对不相关的rtk如可溶性酪氨酸激酶或丝氨酸/苏氨酸激酶的活性较差。s5为实验室前期构建的具有抗血管生成活性的候选化合物,前期研究表明,具有与索拉菲尼相当的vegfr-2抑制活性。
3.蛋白降解靶向嵌合体(proteolysis targeting chimera,protac)是一种能够同时结合靶蛋白和e3泛素连接酶的双功能分子,通过同时结合靶蛋白和e3泛素连接酶,拉近靶蛋白和e3连接酶之间的距离,从而诱导靶蛋白的泛素化,泛素化的靶蛋白能够被26s蛋白酶体识别并降解,达到彻底清除疾病相关蛋白的目的。与小分子抑制剂相比,protac具有用量少,不易产生耐药性等优点,所以在新药研发领域呈现出蓬勃发展的态势。但蛋白降解靶向嵌合体固有的特性——分子量大导致其理化性质及细胞渗透性差,限制了其进一步发展,因此亟需对其药代动力学性质进行优化。
技术实现要素:
4.本发明的目的在于提供一种可细胞内自组装的蛋白降解剂及其制备方法和应用,该制备方法简单,易于实现,并且收率较高,降解剂通过自组装形成,克服传统蛋白降解剂分子量大的固有缺陷,可用于制备抗肿瘤药物。
5.为达到上述目的,本发明采用以下技术方案:
6.一种细胞内自组装蛋白降解剂,该降解剂的结构式如下:
[0007][0008]
其中x=1~6,r1=ch3/cl,r2=f/h,r3为甲基或氢,r4为亚甲基或苯基,r6为亚甲基或羰基,r5为亚甲基或者
[0009]
一种细胞内自组装蛋白降解剂的制备方法,包括以下步骤:
[0010]
使用链接体将靶蛋白配体linifanib或靶蛋白配体s5与降冰片烯连接,获得带有
降冰片烯的靶蛋白配体;其中,靶蛋白配体s5的结构式如下:
[0011][0012]
通过连接链将四嗪基团修饰在e3泛素连接酶配体上,得到带有四嗪的e3泛素连接酶配体;
[0013]
通过分步给药的方法,使得带有降冰片烯的靶蛋白配体与带有四嗪的e3泛素连接酶配体先后进入细胞,在细胞内发生生物正交反应,自组装形成蛋白降解剂。
[0014]
进一步的,带有降冰片烯的靶蛋白配体具体过程以下过程制得:靶蛋白配体linifanib或靶蛋白配体s5与链接体经酰胺缩合反应,得到带有boc保护基化合物,然后经氯化氢的乙酸乙酯作用,脱除boc保护基,再与5-降冰片烯-2-羧酸经酰胺缩合反应,得到带有降冰片烯的靶蛋白配体分子。
[0015]
进一步的,带有降冰片烯的靶蛋白配体的结构式如下:
[0016][0017]
其中,x=1~6,r1=ch3/cl,r2=f/h。
[0018]
进一步的,带有四嗪的e3泛素连接酶配体具体通过以下过程制得:
[0019]
将乙腈、氰基化合物以及水合肼在三氟甲磺酸锌或三氟甲磺酸镍的作用下,或将醋酸甲脒、氰基化合物以及水合肼在催化剂三氟甲磺酸锌的作用下,经环化反应,氧化脱氢,形成带有不同取代基的1,2,4,5-四嗪类化合物,然后在三氟乙酸的作用下脱去boc保护基,再通过连接链与e3泛素连接酶配体沙利度胺类似物连接,形成带有四嗪的e3泛素连接酶配体。
[0020]
进一步的,带有四嗪的e3泛素连接酶配体的结构式如下:
[0021][0022]
其中,r3为甲基或氢,r4为亚甲基或苯基,r6为亚甲基或羰基,r5为亚甲基或者
[0023]
进一步的,蛋白降解剂具体通过以下过程制得:将带有降冰片烯的靶蛋白配体溶液加入到含细胞的培养皿中孵育2h后,再加入带有四嗪的e3泛素连接酶配体溶液,孵育,在细胞内经自组装形成的蛋白降解剂。
[0024]
一种如上所述的细胞内自组装蛋白降解剂在制备抗肿瘤药物中的应用。
[0025]
进一步的,抗肿瘤药物为选择性诱导pdgfr-β蛋白降解的药物。
[0026]
进一步的,抗肿瘤药物为抗神经胶质瘤药物。
[0027]
与现有技术相比,本发明具有以下有益效果:
[0028]
本发明通过构建一类带有生物正交基团的靶向识别分子,通过分步给药使其能够在细胞内自组装形成蛋白降解剂,相比于整体型蛋白降解靶向嵌合体,本发明构建的细胞内自组装型蛋白降解靶向嵌合体,不仅能够利用蛋白降解靶向嵌合体的作用机制降解疾病相关蛋白,还能够以自组装的方式减小化合物的分子量,解决整体蛋白降解剂分子量大的问题,增加细胞渗透性,优化其理化性质,增强作用效果。本发明构建的细胞内自组装形成蛋白降解剂的制备方法简单,易于实现,并且收率较高。
[0029]
本发明的小分子蛋白降解靶向嵌合体能够用于制备治疗癌症的药物中,尤其用于制备以pdgfr-β为靶点的抗肿瘤药物中。
附图说明
[0030]
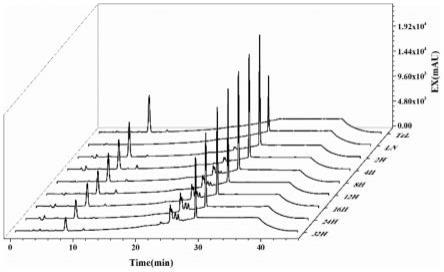
图1为本发明构建的靶向识别分子ln与tzl生物正交反应过程(2-32h)的高效液相色谱图;
[0031]
图2为本发明构建的靶向识别分子ln与tzf生物正交反应过程(2-32h)的高效液相色谱图;
[0032]
图3为本发明构建的靶向识别分子ln与tzl自组装形成的蛋白降解剂对u87细胞的蛋白降解效果考察。其中,a为靶蛋白配体分子ln与带有四嗪标签的e3泛素连接酶配体tzl自组装形成的蛋白降解剂的对蛋白的降解效果,b为靶蛋白配体分子tzl与带有四嗪标签的e3泛素连接酶配体tzl自组装形成的蛋白降解剂的对蛋白的降解效果。
具体实施方式
[0033]
下面结合附图和具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
[0034]
生物正交反应是指能够在活体细胞或组织中发生的、并且能够不干扰生物自身生化反应条件下可以进行的一类化学反应,具有简单、高效、高特异性的特点。研究表明,四嗪可以在无需催化剂的条件下与环状烯烃或炔烃快速高效地发生生物正交反应生成稳定的产物,且此反应对细胞无损害。因此拟将蛋白降解靶向嵌合技术与生物正交反应结合起来,构建带有生物正交基团的、分别靶向靶蛋白和e3泛素连接酶的化合物分子,两部分先后进入细胞,在细胞内发生生物正交反应自组装形成蛋白降解剂,进而发挥降解靶蛋白的作用。基于该策略,可以降低protac分子量,优化其药动药代性质,增加细胞渗透性,以期提高其作用效果。
[0035]
本发明通过使用不同长度的linker将靶蛋白配体linifanib或s5和生物正交基团降冰片烯连接获得带有生物正交基团降冰片烯的靶蛋白配体分子;通过不同类型的连接链将四嗪基团修饰在e3泛素连接酶配体上得到带有生物正交基团四嗪的e3泛素连接酶配体。通过分步给药的方法,使得这二者能够先后进入细胞,在细胞内发生生物正交反应自组装形成蛋白降解剂,从而达到减小分子量,增加细胞渗透性,优化传统蛋白降解剂固有缺陷的目的。本发明中在细胞内自组装形成的蛋白降解剂能够在治疗癌症中应用。本发明涉及的
蛋白降解靶向嵌合体(protacs)能够选择性诱导pdgfr-β蛋白的降解。
[0036]
本发明提供了一种具有抗肿瘤活性的自组装型蛋白降解剂,该蛋白降解剂在体外具有抗肿瘤活性,可应用于抗肿瘤药物的制备。
[0037]
一种细胞内自组装蛋白降解剂,包括带有生物正交基团(降冰片烯)的靶蛋白配体分子和带有生物正交基团(四嗪)的e3泛素连接酶配体分子,所述带有生物正交基团(降冰片烯)的靶蛋白配体分子具有如下结构式:
[0038][0039]
其中,x=1~6,r1=ch3/cl,r2=f/h。
[0040]
所述带有生物正交基团(四嗪)的e3泛素连接酶配体分子具有如下结构式:
[0041][0042]
其中,r3可以为甲基或氢,r4可以为亚甲基或苯基,r6可以为亚甲基或羰基,r5可以为不同长度的连接链,优选的,为亚甲基或
[0043]
如上所述的细胞内自组装形成蛋白降解剂的化合物的制备方法,包括以下合成步骤:
[0044]
1)所述带有生物正交基团(降冰片烯)的靶蛋白配体分子的制备方法如下:
[0045]
抗肿瘤活性分子linifanib或s5与不同长度的linker(链接体,例如boc保护的氨基丁酸)经酰胺缩合反应,柱色谱纯化得到一类带有boc保护基化合物,然后经氯化氢的乙酸乙酯作用脱除boc保护基暴露出活性反应基团氨基,再与5-降冰片烯-2-羧酸经酰胺缩合反应得到带有生物正交基团(降冰片烯)的靶蛋白配体分子。
[0046]
2)带有生物正交基团(四嗪)的e3泛素连接酶配体分子的制备方法如下:
[0047]
乙腈或醋酸甲脒、不同类型的氰基化合物以及水合肼在催化剂三氟甲磺酸锌或三氟甲磺酸镍的作用下,经环化反应,氧化脱氢形成带有不同取代基的1,2,4,5-四嗪类化合物,而后在三氟乙酸的作用下脱去boc保护基暴露出氨基活性基团,再通过不同类型的连接链与e3泛素连接酶配体沙利度胺类似物连接在一起形成带有生物正交基团(四嗪)的e3泛素连接酶配体分子。
[0048]
将带有生物正交基团(降冰片烯)靶蛋白配体分子加入到含肿瘤细胞的培养皿中孵育2h后,再加入相同浓度的带有生物正交基团(四嗪)的e3泛素连接酶配体分子,置于37℃,5%co2恒温培养箱中孵育48h,在细胞内经自组装形成的蛋白降解剂的结构,其结构通式如下:
[0049][0050]
其中x=1~6,r1=ch3/cl,r2=f/h,r3可以为甲基或氢,r4可以为亚甲基或苯基,r5可以为不同长度的连接链,r6可以为亚甲基或羰基。
[0051]
所述蛋白降解剂的化合物在制备抗肿瘤药物中的应用。
[0052]
抗肿瘤药物为抗神经胶质瘤药物。
[0053]
下面结合图中所示的合成路线和具体的合成实施例来详细说明本发明提供的一类用于细胞内自组装形成蛋白降解剂的靶向识别分子的制备和活性筛选方法。
[0054]
实施例1
[0055]
一种带有生物正交基团降冰片烯的靶蛋白配体分子ln的制备方法,包括以下合成步骤:
[0056]
1)将48.5mmolγ-氨基丁酸溶于80ml的四氢呋喃并置于冰水浴中,加入1mol/l的氢氧化钠溶液80ml,然后逐滴滴加53.3mmol二碳酸二叔丁酯的四氢呋喃溶液,并室温搅拌,茚三酮检测反应进程,待反应完毕后,减压旋除可挥发性溶剂,用1mol/l盐酸调节至2~3,乙酸乙酯萃取,饱和氯化钠洗涤有机相,无水硫酸钠干燥,抽滤,减压旋除溶剂即得浅黄色化合物1,结构式如下,lc-ms(esi,m/z):204.30[m+h]
+
,202.10[m+h]
+
。
[0057][0058]
2)将0.80mmol化合物1,0.60mmol hatu溶于干燥二氯甲烷中,在冰浴条件下,滴加1.60mmol dipea,室温搅拌15min,加入0.40mmol linifanib,室温搅拌过夜,加水,二氯甲烷萃取,饱和氯化钠洗涤有机相,无水na2so4干燥,抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得透明油状物质,即化合物2(0.22g),结构式如下,产率为98.30%,lc-ms(esi,m/z):561.25[m+na]
+
,559.15[m-h]-。
[0059][0060]
3)将0.39mmol化合物2溶于2mol/l氯化氢的乙酸乙酯溶液中,室温搅拌过夜,抽滤所得滤饼(白色固体)即为化合物3(0.18g),结构式如下,产率为99.61%,lc-ms(esi,m/z):
461.15[m+h]
+
,459.15[m-h]-。
[0061][0062]
4)将0.47mmol 5-降冰片烯-2-羧酸,0.70mmol hatu溶于干燥二氯甲挖中,在冰浴条件下,逐滴加入1.86mmol dipea,搅拌10min后,加入0.47mmol化合物3,室温搅拌8h。反应完毕后,旋除有机相,加入适量的水,乙酸乙酯萃取(3
×
),饱和氯化钠洗涤,无水na2so4干燥。抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得白色产物,即带有生物正交基团降冰片烯的靶蛋白配体分子ln(90mg),结构式如下,产率为33.33%,lc-ms(esi,m/z):581.25[m+h]
+
,579.15[m-h]-。
[0063][0064]
实施例2
[0065]
一种带有生物正交基团降冰片烯的靶蛋白配体分子s5n的制备方法,包括以下合成步骤:
[0066]
1)将48.5mmolγ-氨基丁酸溶于80ml的四氢呋喃并置于冰水浴中,加入1mol/l的氢氧化钠溶液80ml,然后逐滴滴加53.3mmol二碳酸二叔丁酯的四氢呋喃溶液,并室温搅拌,茚三酮检测反应进程,待反应完毕后,减压旋除可挥发性溶剂,用1m hcl调节至2~3,乙酸乙酯萃取,饱和氯化钠洗涤有机相,无水硫酸钠干燥,抽滤,减压旋除溶剂即得浅黄色化合物1,结构式如下,lc-ms(esi,m/z):204.30[m+h]
+
,202.10[m+h]
+
。
[0067][0068]
2)将0.794mmol化合物1,0.794mmol hatu溶于干燥二氯甲烷中,在冰浴条件下,滴加1.588mmol dipea,室温搅拌15min,加入0.397mmol s5,室温搅拌过夜,加水,二氯甲烷萃取,饱和氯化钠洗涤有机相,无水na2so4干燥,抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得透明油状物质,即化合物4(0.22g),结构式如下,产率为98.65%,lc-ms(esi,m/z):585.25[m+na]
+
,561.15[m-h]-。
[0069][0070]
3)将0.391mmol化合物4溶于2mol/l氯化氢的乙酸乙酯溶液中,室温搅拌过夜,抽滤所得滤饼(白色固体)即为化合物5(0.18g),结构式如下,产率为99.44%,lc-ms(esi,m/z):463.10[m+h]
+
,461.05[m-h]-。
[0071][0072]
4)将0.389mmol 5-降冰片烯-2-羧酸,0.584mmol hatu溶于干燥二氯甲挖中,在冰浴条件下,逐滴加入1.558mmol dipea,搅拌20min后加入0.389mmol化合物5,室温搅拌8h。反应完毕后,旋除有机相,加入适量的水,乙酸乙酯萃取(3
×
),饱和氯化钠洗涤,无水na2so4干燥。抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得白色产物,即带有生物正交基团降冰片烯的靶蛋白配体分子s5n(94mg),结构式如下,产率为41.46%,lc-ms(esi,m/z):583.15[m+h]
+
,581.10[m-h]-。
[0073][0074]
将实施例1中的γ-氨基丁酸改为7-氨基庚酸即可制备带有生物正交基团降冰片烯的靶蛋白配体l7n(x=4),具有如下结构式:
[0075][0076]
将实施例1中的γ-氨基丁酸改为9-氨基壬酸即可制备带有生物正交基团降冰片烯的靶蛋白配体l9n(x=6),具有如下结构式:
[0077][0078]
实施例3
[0079]
一种带有生物正交基团四嗪的e3泛素连接酶配体tzl的制备方法,包括以下合成
步骤:
[0080]
1)在氮气保护下,25mmol乙腈、2.5mmol(4-氰基苄基)氨基甲酸叔丁酯、1.25mmol三氟甲磺酸锌以及125mmol(6.067ml)的80%水合肼(质量分数)混合置于60℃反应36h,反应液冷却至室温,将50mmol亚硝酸钠溶液(3.45g亚硝酸钠溶于20ml的水)加入反应液中,然后缓慢滴加1mol/l盐酸直到无气泡产生且ph为3。乙酸乙酯萃取2次,合并有机相,无水na2so4干燥。抽滤除去干燥剂,经柱层析分离得到紫红色粉末,即化合物6(0.11g),结构式如下,产率为14.67%,lc-ms(esi,m/z):302.40[m+h]
+
。
[0081][0082]
2)将0.43mmol化合物3溶于4ml干燥二氯甲烷中,在冰浴条件下滴加1ml三氟乙酸,室温搅拌2h,直接旋干,得到化合物7,快速用于下一步反应,将化合物7与0.43mmol丁二酸酐混溶于10ml干燥二氯甲烷溶液中,加入200μl dmf助溶,在冰浴条件下逐滴加入1.73mmol dipea,室温反应过夜。反应结束后,减压旋除二氯甲烷,经柱色谱分离得红色粉末,即化合物8(0.12g),结构式如下,产率为93.02%,lc-ms(esi,m/z):302.30[m+h]
+
。
[0083][0084]
3)将48.5mmolγ-氨基丁酸溶于80ml的四氢呋喃并置于冰水浴中,加入1mol/l的氢氧化钠溶液80ml,然后逐滴滴加53.3mmol二碳酸二叔丁酯的四氢呋喃溶液,并室温搅拌,茚三酮检测反应进程,待反应完毕后,减压旋除可挥发性溶剂,用1mol/l盐酸调节至2~3,乙酸乙酯萃取,饱和氯化钠洗涤有机相,无水硫酸钠干燥,抽滤,减压旋除溶剂即得浅黄色化合物1(8.24g),结构式如下,产率为83.65%,lc-ms(esi,m/z):204.30[m+h]
+
,202.10[m+h]
+
。
[0085][0086]
4)将0.85mmol化合物1与1.16mmol hatu溶于6ml dmf中,在冰浴条件下,滴加1.54mmol三乙胺,室温搅拌15min,加入0.77mmol来那度胺(lenalidomide),室温搅拌过夜,加水,乙酸乙酯萃取,饱和氯化钠洗涤有机相,无水na2so4干燥,抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得透明油状物质,即化合物9(0.42g),结构式如下,产率为97.66%,lc-ms(esi,m/z):467.10[m+na]
+
,443.05[m-h]-。
[0087][0088]
5)将0.94mmol化合物9溶于2mol/l氯化氢的乙酸乙酯溶液中,室温搅拌2h,抽滤所得滤饼(白色固体)即为化合物10(0.32g),结构式如下,产率为99.76%,lc-ms(esi,m/z):345.05[m+h]
+
,342.90[m-h]-。
[0089][0090]
6)将0.42mmol化合物8与0.71mmol hatu溶于dmf中,在冰浴条件下,逐滴加入1.9mmol dipea,搅拌15min后,加入0.42mmol化合物10,室温搅拌8h。反应完毕后,加入适量的水,二氯甲烷萃取(3
×
),无水na2so4干燥。抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得红色产物tzl(75mg),结构式如下,产率为28.52%,lc-ms(esi,m/z):650.65[m+na]
+
,626.15[m-h]-。
[0091][0092]
实施例4
[0093]
一种带有生物正交基团四嗪的e3泛素连接酶配体tzf的制备方法,包括以下合成步骤:
[0094]
1)在氮气保护下,25mmol乙腈、2.5mmol(4-氰基苄基)氨基甲酸叔丁酯、1.25mmol三氟甲磺酸锌以及125mmol(6.067ml)的80%水合肼(质量分数)混合置于60℃反应36h,反应液冷却至室温,将50mmol亚硝酸钠溶液(3.45g亚硝酸钠溶于20ml的水)加入反应液中,然后缓慢滴加1mol/l盐酸直到无气泡产生且ph为3。乙酸乙酯萃取2次,合并有机相,无水na2so4干燥。抽滤除去干燥剂,经柱层析分离得到紫红色粉末,即化合物6(0.11g),结构式如下,产率为14.67%,lc-ms(esi,m/z):302.40[m+h]
+
。
[0095][0096]
2)将0.63mmol化合物6溶于3ml干燥二氯甲烷中,在冰浴条件下滴加1ml三氟乙酸,室温搅拌2h,直接旋干,得到化合物7,结构式如下,快速用于下一步反应。
[0097][0098]
3)将1.81mmol化合物thalidomide fluoride,2.00mmol甘氨酸叔丁酯盐酸盐,2.72mmol dipea,4ml dmf混溶于100ml茄形瓶中,置于微波反应仪中于85℃下反应50min,反应完毕后,加水,乙酸乙酯萃取,饱和氯化钠洗涤,无水na2so4干燥。抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得黄色荧光物质,即化合物11(0.48g),结构式如下,产率为68.57%,lc-ms(esi,m/z):410.05[m+na]
+
,386.00[m-h]-。
[0099][0100]
4)将0.93mmol化合物11溶于6ml干燥二氯甲烷中,在冰浴条件下,逐滴加入2ml三氟乙酸,室温搅拌过夜,减压旋干,经柱色谱分离得黄色荧光产物,即为化合物12(0.11g),结构式如下,产率为35.48%。lc-ms(esi,m/z):331.00[m+h]
+
,329.90[m-h]-。
[0101][0102]
5)将0.63mmol化合物12与0.94mmol hatu溶于dmf中,在冰浴条件下,逐滴加入2.51mmol dipea,搅拌10min后,加入0.63mmol化合物7,室温搅拌8h。反应完毕后,加入适量的水,二氯甲烷萃取(3
×
),无水na2so4干燥。抽滤除去干燥剂,减压旋除溶剂,经柱色谱分离得带有黄色荧光的红色产物,即带有生物正交基团四嗪的e3泛素连接酶配体tzf(40mg),结构式如下,产率为12.51%,lc-ms(esi,m/z):515.05[m+h]
+
,513.05[m-h]-。
[0103][0104]
实施例5
[0105]
带有生物正交基团(降冰片烯)的靶蛋白配体分子与带有生物正交基团(四嗪)的e3泛素连接酶配体分子在细胞内经自组装形成的蛋白降解剂
[0106]
1)生物正交反应生成蛋白降解剂
[0107]
采用高效液相色谱在体外监测两部分靶向识别分子能否发生生物正交反应、反应速率以及反应进程。相同浓度的两部分靶向识别分子置于pbs和乙腈(v:v=1:1)混合体系中按体积比(1:1)混匀,置于37℃恒温摇床上反应,在不同时间点取样进行检测,结果如图1~2所示,靶向识别分子能够互相发生生物正交反应形成蛋白降解剂。
[0108]
2)细胞内自组装生成蛋白降解剂
[0109]
先用带有生物正交基团(降冰片烯)靶蛋白配体分子(终浓度为10μm)处理肿瘤细胞2h后,再给予肿瘤细胞带有生物正交基团(四嗪)的e3泛素连接酶配体分子(终浓度为10μm),置于37℃,5%co2恒温培养箱中孵育48h,然后用pbs洗涤,洗涤后将细胞消化离心收集,最后用细胞破碎仪将细胞破碎,并通过滤膜过滤,再进行电喷雾质谱测定,得到细胞内自组装降解剂产物。
[0110]
实施例6
[0111]
带有生物正交基团降冰片烯的靶蛋白配体分子的细胞增殖抑制活性测定。
[0112]
带有生物正交基团降冰片烯的靶向识别分子ln和s5n在细胞水平的活性检测采用mtt检测法。将处于对数增长期u87细胞,用0.25%胰蛋白酶消化,制成单细胞悬液,接种于96孔板(4000个/孔),每孔180μl。放入37℃,5%co2恒温培养箱中培养,24h后待细胞贴壁后加药。每组设置4个复孔,阴性对照组与空白组均加入20μl/孔无血清培养基,实验组加入不同浓度的药物20μl/孔(以无血清培养基稀释药物),放入37℃,5%co2恒温培养箱中继续培养。药物作用48h后,加入mtt溶液(终浓度为0.5mg/ml)22μl/孔,37℃孵育4h后,小心吸弃上清液,加入dmso 150μl/孔,置于摇床上充分振摇15min。用酶联免疫检测仪于490nm波长下测定各孔吸光度(od)值。
[0113]
数值处理:抑制率=(od
阴性组-od
给药组
)/(od
阴性组-od
空白组
)
×
100%;
[0114]
部分化合物的实验结果见表1:
[0115]
表1靶向识别分子的细胞增殖抑制活性
[0116][0117]
从表1可以看出,相比于母体化合物本身,本发明制备靶向识别分子s5n对u87细胞具有较好的抑制活性。
[0118]
实施例7
[0119]
细胞内自组装型蛋白降解剂对靶蛋白降解效果的考察。
[0120]
将处于对数增长期的u87细胞,用0.25%胰蛋白酶消化,制成单细胞悬液,接种于6
孔板(5
×
105个/孔),每孔2ml。放入37℃,5%co2恒温培养箱中培养,24h后待细胞贴壁后加药。先用固定浓度(终浓度为10μm)或不同浓度(终浓度为0.016μm,0.08μm,0.4μm,2μm,10μm)的靶蛋白配体分子处理细胞2h后,再给予不同浓度(终浓度为0.016μm,0.08μm,0.4μm,2μm,10μm)或者固定浓度(终浓度为10μm)的带有四嗪标签的e3泛素连接酶配体处理细胞,置于37℃,5%co2恒温培养箱中孵育48h,然后进行蛋白提取,再采用western blot免疫印迹法检测相关蛋白水平,参见图3中a和b,结果显示,由靶蛋白配体分子ln与带有四嗪标签的e3泛素连接酶配体tzl自组装形成的蛋白降解剂的对蛋白的降解效果不佳,并未达到预期,可能是由于生物正交基团的引入,化合物的活性降低,并且这两者发生生物正交的反应速率也不够迅速,可能也是引起其活性不佳的原因之一。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1