RNA靶向测序基因芯片的质控方法及应用与流程
rna靶向测序基因芯片的质控方法及应用
技术领域
1.本发明涉及基因芯片的质控方法,更具体地,本发明涉及一种用于rna靶向测序的液相基因芯片的质控方法及应用。
背景技术:
2.基因芯片(gene panel)是由生物素修饰的探针组成,用于富集基因组目标区域序列,通常与高通量测序结合,具有准确性高,经济、通量高等优势。基因芯片,特别是液相基因芯片已经被广泛用于基因组单核苷酸变异(snv)、拷贝数变异(cnv)等研究。
3.除被用于基因组的靶向区域富集外,基因芯片也被用于rna的靶向测序(rna-cap),并进行相关的序列变异研究,例如snv和基因融合变异(gene fusion)。在融合基因的检测中,rna-cap不需融合基因的先验知识,能够对未知融合亚型进行检测。同时,rna-cap可以一次完成对成百上千个基因的检测,通量优势明显。再结合高深度测序,可以保证检测的灵敏度。因此,用于rna-cap的液相基因芯片在rna序列变异研究中扮演着重要角色。
4.但是由于不同基因的表达量差异,rna-cap数据中,不同基因的覆盖深度存在差异,低表达量基因往往覆盖深度较低。因此,如何判断rna-cap的基因芯片中,针对低表达量基因的探针是否有效工作,是验证rna-cap基因芯片的难题。
5.背景技术中的信息仅仅在于说明本发明的总体背景,不应视为承认或以任何形式暗示这些信息构成本领域一般技术人员所公知的现有技术。
技术实现要素:
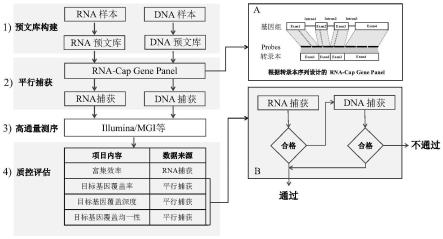
6.本发明利用dna与rna样本进行平行捕获,通过dna和rna样本的捕获数据综合评估rna靶向测序的基因芯片的性能,有效解决了由于rna样本中基因表达差异等造成的对rna靶向测序的液相基因芯片捕获性能无法准确评估的问题。具体地,本发明包括以下内容。
7.本发明的第一方面,提供一种rna靶向测序基因芯片的质控方法,其包括以下步骤:
8.(1)分别提供dna预文库和rna预文库;
9.(2)获得第一测序数据和第二测序数据的步骤,其中,第一测序数据为利用基因芯片对所述dna预文库进行杂交捕获及测序而得到数据,第二测序数据为利用基因芯片对所述rna预文库进行平行杂交捕获及测序而得到的数据,其中所述基因芯片包括针对不同目标片段的多条探针;和
10.(3)利用所述第一测序数据和所述第二测序数据的参数来评估所述基因芯片的性能。
11.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,所述基因芯片为液相基因芯片。
12.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,参数包括富集效率、基因覆盖率、覆盖深度和均一性中的至少一种。
13.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,当第一测序数据中目标片段的覆盖率和覆盖深度分别大于规定阈值,而第二测序数据所述目标片段的覆盖率和覆盖深度分别小于规定阈值时,则所述目标片段对应的探针工作有效。
14.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,当第一测序数据中目标片段的覆盖率和/或覆盖深度以及第二捕获数据中所述目标片段的覆盖率和/或覆盖深度均小于规定阈值时,则将所述目标片段对应的探针认定为工作无效。
15.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,所述dna预文库和所述rna预文库分别来自于同一样本或不同样本。
16.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,所述目标片段为基因内部的不同片段,所述参数包括均一性。
17.在某些实施方案中,根据第一方面所述的rna靶向测序基因芯片的质控方法,其中,所述目标片段为不同基因,所述参数包括覆盖率和覆盖深度。
18.本发明的第二方面,提供一种液相基因芯片优化方法,其包括:
19.(a)根据第一方面所述的rna靶向测序基因芯片的质控方法来确定所述液相基因芯片的目标片段中的黑名单的步骤;和
20.(b)对于黑名单目标基因进一步设计优化探针的步骤。
21.本发明的第三方面,提供一种用于rna靶向测序的液相基因芯片,其包括针对不同目标片段的探针,和用于显示或指示目标片段中的黑名单的说明。
22.本发明解决了rna靶向测序的液相基因芯片对低表达量基因的捕获性能无法验证的问题。更具体地,本发明通过rna靶向测序基因芯片对dna样本进行捕获测序及评价,完善利用rna样本进行基因芯片验证的漏洞,具有准确、通用的优势。
附图说明
23.图1为示例性说明rna靶向测序的液相基因芯片质控流程的示意图;
24.图2为全转录组测序与全基因组测序的基因覆盖率和覆盖深度评估;
25.图3为全转录组测序与全基因组测序的覆盖异常基因数量统计;
26.图4为rna和dna样本综合评估rna靶向测序的液相基因芯片原理示意图。相较于转录本存在的基因表达差异,基因组层面的绝大部分基因不存在拷贝数干扰,因此,在检测rna样本的同时,对dna样本也平行开展rna-cap panel捕获,在理论上可以解决rna样本的部分基因由于表达差异和可变剪接等生物现象造成的基因芯片评估干扰。
具体实施方式
27.现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
28.应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为具体公开了该范围的上限和下限以及它们之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围
内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
29.除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。除非另有说明,否则“%”为基于重量的百分数。
30.本文中,术语“基因芯片”是指通过化学修饰使探针固定于固相载体表面得到的探针阵列。
31.本文中,术语“液相基因芯片”也称为微球体悬浮芯片,是由大小均一的圆形微球为主要基质构成的液相体系。其中,每种微球上固定有不同的探针。
32.本文中,术语“探针”是指寡核苷酸分子,特别是根据目标片段的转录本设计的单链dna(ssdna)探针。芯片通常包括多条探针,从而组成探针组。探针组中各探针排布为一重或多重覆盖目标区域。探针的长度不特别限定,一般为90-150nt,优选100-140nt,如110nt、120nt、130nt等。探针组中各探针的长度可以相同,也可以不同。
33.本文中,术语“rna靶向测序基因芯片”是指芯片中的探针是根据目标片段的转录本设计的单链dna探针。
34.本文中,术语“相对覆盖深度”是指在多个目标片段组成组合中,某一目标片段的覆盖深度除以所有目标片段的覆盖深度的平均值得到的数值。例如,当目标片段为基因时,某基因的覆盖深度为100x,而所有目标基因的覆盖深度的平均值为100x时,则此时相对覆盖深度为1。而当某基因的覆盖深度为20x时,则此时相对覆盖深度为0.2。在某些实施方案中,目标片段为基因内部含特定位点的片段。
35.本文中,术语“覆盖率”是指对于规定的序列区域内,覆盖深度大于等于1x的碱基占所述规定的序列区域内所有碱基的比例。例如,某序列区域由100bp序列组成,统计100bp内每个碱基的覆盖深度,将大于等于1x的所有碱基数除以100bp碱基得到的比值为覆盖率。
36.本发明的rna靶向测序基因芯片的质控方法,用于评估基因芯片特别是液相基因芯片的捕获性能,特别是用于评估由于转录引起的变化的准确性。本发明质控方法包括但不限于以下三个步骤:
37.(1)分别提供dna预文库和rna预文库;
38.(2)获得第一测序数据和第二测序数据的步骤;和
39.(3)利用所述第一测序数据和所述第二测序数据的参数来评估所述基因芯片的性能。
40.本发明中,步骤(1)为分别提供dna预文库和rna预文库的步骤,可以包括构建预文库的步骤,也可以直接调用预先自己或由第三方构建的预文库的步骤。dna预文库和rna预文库可以同时构建或先后依次构建,对此不特别限定。预文库构建时的生物样本不特别限定,优选来源于同一生物的样本,如体液,包括血液或其成分、组织液、唾液等;也可是所需组织的样本。dna预文库和rna预文库构建时的生物样本优选为同一样本或相同样本,如相同类型的组织或细胞。
41.本发明中,步骤(2)为获得测序数据的步骤,优选包括利用基因芯片进行捕获和测
序的步骤,如利用基因芯片对所述dna预文库进行杂交捕获及测序,得到第一测序数据,利用基因芯片对所述rna预文库进行平行杂交捕获及测序,得到第二测序数据。其中,基因芯片包括针对不同目标片段的多条探针。基因芯片中探针所对应的目标片段的长度不特别限定。在某些实施方案中,目标片段的长度为1mb以下,例如8000bp以下、6000bp以下、5000bp以下或3000bp以下,另一方面优选为300bp以上、500bp以上,优选1000bp以上。在某些实施方案中,目标片段的长度为1mb以上,例如,2mb以上、3mb以上、4mb以上或5mb以上。
42.本发明的基因芯片所对应的目标片段的数量不特别限定。在某些实施方案中,目标片段的数量为550以下。例如500以下、400以下、300以下、200以下或100以下。在某些实施方案中,目标片段的数量为550以上,例如600以上、700以上、1000以上、5000以上、8000以上,甚至10000以上。
43.本发明中,步骤(3)为基因芯片的性能评估步骤,包括利用第一测序数据和第二测序数据的参数来评估所述基因芯片的性能。其中参数包括富集效率、基因覆盖率、覆盖深度和均一性中的至少一种指标。优选地,本发明的参数包括两种以上的指标,例如基因覆盖率和覆盖深度组合的指标等。各指标可以设定或规定相应阈值。当高于规定的阈值时将对应的指标认定为符合相应要求,进而用于评估。同时还进一步包括将符合相应要求的来源于第二测序数据的指标与来源于第一测序数据的指标相比较的步骤。
44.在某些实施方案中,本发明的评估步骤(3)包括以下判断步骤:当第一测序数据中目标片段的测序参数以及第二捕获数据中目标片段的测序参数均小于规定阈值时,则将目标片段对应的探针认定为工作无效。此时的测序参数包括单独的覆盖率,或者单独的覆盖深度,或者覆盖率和覆盖深度的组合。通过该判断步骤认定的探针或其对应的目标片段可列为基因芯片的黑名单。
45.在某些实施方案中,本发明的评估步骤(3)包括以下判断步骤:当第一测序数据中目标片段的覆盖率和覆盖深度分别大于规定阈值,而第二测序数据所述目标片段的覆盖率和覆盖深度分别小于规定阈值时;或者当第一测序数据中目标片段的覆盖率和覆盖深度分别大于规定阈值,同时第二测序数据所述目标片段的覆盖率和覆盖深度分别大于规定阈值时,则目标片段对应的探针工作有效。
46.需要说明的是,虽然以上为了说明目的而按步骤(1)-(3)顺序详细说明了各步骤,但本领域技术人员已知,在不脱离本发明精神的情况下,可以对步骤(1)-(3)进行调整。例如对于第一测序数据和第二测序数据而言,可以同时或先后获取。在某些实施方案中,本发明的方法包括首先构建dna预文库,接下来进行后续捕获和测序获得第一测序数据。然后,构建rna预文库,接下来进行后续捕获和测序获得第二测序数据。
47.实施例
48.以下实施例使用针对98个基因作为靶基因组作为示例说明液相基因芯片的质控方法。本实施例仅为示例性说明目的,并不意欲限制本发明的范围。
49.实施例1
50.一、98gene rna-cap gene panel的探针设计及合成:
51.对98个基因按照cds(转录本)序列设计探针,探针设计按照1重目标区域覆盖原则,使用5’生物素修饰的120nt单链dna探针,对目标序列进行平铺,探针头尾顺次排列。整个panel覆盖0.23mb区域,探针由伯科生物科技有限公司合成。98个基因名称即转录本信息
如表1所示。
52.表1.98gene rna-cap gene panel基因信息
[0053][0054][0055]
二、rna预文库构建:
[0056]
对rna样本构建两个rna预文库(rna-重复1#、rna-重复2#),rna样本信息:ffpe肿瘤野生型标准品(供应商:菁良;货号:gw-opsm005)。
[0057]
2.1一链合成(供应商:abclonal;货号:rk20353)
[0058]
如表2所示,取100ng总rna加入2x frag/elution buffer,涡旋混匀后,短暂离心,然后在pcr仪上85℃孵育10min(热盖105℃)。
[0059]
表2
[0060]
组分体积(μl)100ng总rna5.02xfrag/elutionbuffer5总体积10
[0061]
如表3所示,在冰上向上一步产物中加入rt reagent和first strand synthesis enzyme mix,涡旋混匀后,短暂离心。
[0062]
表3
[0063]
组分体积(μl)rna打断产物10rtreagent8firststrandsynthesisenzymemix2总体积20
[0064]
设置pcr程序(表4),将配制好的反应液放入,运行程序(热盖105℃)。
[0065]
表4
[0066][0067]
2.2二链合成(供应商:abclonal;货号:rk20346)
[0068]
如表5所示,在冰上向上一步产物中加入second strand synthesis reaction buffer、second strand synthesis enzyme mix和无酶无菌水,涡旋混匀后,短暂离心。将配制好的反应液放入pcr仪,在16℃条件下孵育1h(热盖关闭)。
[0069]
表5
[0070]
组分体积(μl)一链合成产物20secondstrandsynthesisreactionbuffer8secondstrandsynthesisenzymemix4无酶无菌水48总体积80
[0071]
2.3二链合成产物纯化
[0072]
反应结束后,向反应液中加入144μl vahts dna clean beads(供应商:诺唯赞;货号:n411-03),充分涡旋混匀后,室温孵育5min,短暂离心后置于磁力加上吸附5min,吸弃上清,加入180μl 80%乙醇孵育30sec后,吸弃上清,再加入180μl 80%乙醇孵育30sec后,吸弃上清。将反应管短暂离心,置于磁力架上,吸弃残留液体,室温干燥。5min后,加入52μl low-te缓冲液,涡旋混匀,室温静置2min,短暂离心后,置于磁力架上,吸附2min,将50μl上清液转移至新的pcr管中。
[0073]
2.4末端修复
[0074]
如表6所示,向二链合成产物中加入end prep mix4(供应商:诺唯赞;货号:nd607-02),涡旋混匀后,短暂离心。将配制好的反应液放入pcr仪,按照表7所示程序运行(热盖75℃)。
[0075]
表6
[0076]
组分体积(μl)二链合成产物50endprepmix415
总体积65
[0077]
表7
[0078][0079]
2.5接头连接
[0080]
反应结束后,向上一步产物中加入表8所示试剂(供应商:诺唯赞;货号:nd607-02),涡旋混匀后,短暂离心,在20℃条件下孵育30min。
[0081]
表8
[0082]
组分体积(μl)末端修复产物65illumina接头vazyme-s2.5rapidligationbuffer225rapiddnaligase5无酶无菌水2.5总体积100
[0083]
2.6接头连接产物纯化
[0084]
反应结束后,向反应液中加入90μl vahts dnaclean beads(供应商:诺唯赞;货号:n411-03),充分涡旋混匀后,室温孵育5min,短暂离心后置于磁力加上吸附5min,吸弃上清,加入180μl 80%乙醇孵育30sec后,吸弃上清,再加入180μl 80%乙醇孵育30sec后,吸弃上清。将反应管短暂离心,置于磁力架上,吸弃残留液体,室温干燥。5min后,加入22μl low-te缓冲液,涡旋混匀,室温静置2min,短暂离心后,置于磁力架上,吸附2min,将20μl上清液转移至新的pcr管中。
[0085]
2.7 index pcr扩增
[0086]
向20μl接头连接产物中加入25ul vahts hifi amplification mix和5ul index primer(供应商:诺唯赞;货号:n411-03),按照表9程序进行pcr扩增。
[0087]
表9
[0088][0089]
2.8rna预文库纯化
[0090]
反应结束后,向反应液中加入45μl vahts dna clean beads(供应商:诺唯赞;货号:n411-03)进行纯化,纯化操作同“2.6”步骤。5min后,加入42μl无酶无菌水,涡旋混匀,室温静置2min,短暂离心后,置于磁力架上,吸附2min,将40μl上清液转移至新的pcr管中。
[0091]
2.9 rna预文库质检
[0092]
使用qubit荧光计3.0(thermofisher)测量文库浓度。使用agilent 2100测量文库片段长度,产物主带集中在~300bp,无接头二聚。
[0093]
三、rna预文库与98gene rna-cap gene panel杂交捕获:
[0094]
按照步骤如a-j所示进行16小时杂交捕获。
[0095]
a.文库预封闭
[0096]
将表10试剂加入到0.2ml低吸附离心管(eppendorf)中,使用真空浓缩仪(eppendorf)将离心管中溶液蒸干备用。
[0097]
表10
[0098][0099]
b.探针与文库杂交
[0100]
将13μl杂交缓冲液(0.33m sodium phosphate buffer ph7.0、0.65%sds(w/v)、1.31mm edta、1.31x ssc、2.62x denhardt’s solution、20%甲酰胺(v/v))加入到上述步骤的离心管中,涡旋混匀,室温孵育5分钟。
[0101]
95℃变性10分钟,随后加入4μl 98gene rna-cap gene panel(0.4fmol/probe/rxn),涡旋混匀,65℃孵育16小时。
[0102]
c.清洗液准备
[0103]
按照表11所示准备清洗缓冲液,其中,1x wash buffer s和部分1x wash buffer i在65℃条件下预热30分钟后使用。
[0104]
表11
[0105][0106]
1x beads wash buffer:1m nacl、10mm tris-hcl ph 7.5、1mm edta、0.1%(v/v)tween-20
[0107]
1x wash buffer s:1x ssc、0.1%(v/v)tween-20,ph 7.0
[0108]
1x wash buffer i:1x ssc、0.1%(w/v)sds,ph 7.0
[0109]
1x wash buffer ii:0.5x ssc,ph 7.0
[0110]
1x wash buffer iii:0.2x ssc,ph 7.0
[0111]
d.链霉亲和素磁珠准备
[0112]
将链霉亲和素磁珠(dyna beads m270,invitrogen)从冰箱中(4℃)取出恢复到室温(约30分钟)。涡旋混匀15秒。取100μl链霉亲和素磁珠加入到新的1.5ml低吸附离心管中。将离心管放到磁力架上,直到溶液澄清。吸弃上清,切勿扰动磁珠。按以下步骤对链霉亲和
素磁珠进行清洗:
[0113]
(1)将离心管从磁力架上取下,加入200μl 1x beads wash buffer,涡旋振荡10秒。
[0114]
(2)将离心管瞬时离心,放到磁力架上,直到溶液澄清,吸弃上清,切勿扰动磁珠。
[0115]
重复步骤(1)和(2)。
[0116]
将离心管从磁力架上取下,加入100μl 1x beads wash buffer。将离心管中的100μl磁珠重悬液转移到新的0.2ml低吸附离心管(eppendorf)中待用。将离心管放到磁力架上,直到溶液澄清。吸弃上清,切勿扰动磁珠,立即进行后续实验步骤。
[0117]
e.链霉亲和素磁珠捕获
[0118]
将杂交混合物加入到含链霉亲和素磁珠的0.2ml低吸附离心管中。使用移液器轻柔吹吸10次混匀。使用pcr仪(热盖温度设置为75℃)65℃孵育45分钟。每12分钟涡旋混匀3秒,确保磁珠处于悬浮状态。
[0119]
f.捕获后清洗
[0120]
1.65℃清洗步骤:
[0121]
将100μl预热的1x wash buffer i加入到含有杂交混合物的0.2ml低吸附离心管中。吹吸混匀后,将含有链霉亲和素磁珠的反应液转移到新的1.5ml低吸附离心管中。将离心管放置到磁力架上,直到溶液澄清,吸弃上清。
[0122]
继续按以下步骤进行清洗:
[0123]
(1)加入200μl预热的1x wash buffer s,吹吸或涡旋混匀后,在65℃条件下孵育5分钟。
[0124]
(2)瞬时离心,将离心管放置到磁力架上,直到溶液澄清,吸弃上清。
[0125]
重复步骤(1)和(2)。
[0126]
2.室温清洗
[0127]
加入200μl1x wash buffer i,涡旋混匀2分钟。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。加入200μl1x wash buffer ii,涡旋混匀1分钟。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。加入200μl1x wash buffer iii,涡旋混匀30秒。将离心管瞬时离心,放置到磁力架上,直到溶液澄清,吸弃上清。
[0128]
3.磁珠重悬
[0129]
立即加入20μl无酶无菌水。使用移液器吹吸10次,重悬磁珠,进入后续实验步骤。
[0130]
g.pcr扩增
[0131]
按照表12配制pcr反应体系。
[0132]
表12
[0133][0134]
吹吸或低速涡旋混匀使磁珠保持悬浮状态,立即进入pcr步骤。使用pcr仪按表13
程序运行,热盖温度105℃。
[0135]
表13
[0136][0137]
h.pcr产物纯化
[0138]
每个pcr管中加入75μl vahts dnaclean beads(供应商:诺唯赞;货号:n411-03)。纯化操作如“2.6”步骤所述。使用22μl tris-hcl(10mm,ph8.5)进行洗脱。转移20μl包含捕获文库的洗脱液到新的1.5ml低吸附离心管(eppendorf)中。
[0139]
i.文库质控
[0140]
使用qubit荧光计3.0(thermofisher)测量文库浓度。使用agilent 2100测量文库片段长度,产物主带集中在320bp,无接头二聚。
[0141]
四、高通量测序
[0142]
将捕获文库采用illumina novaseq6000测序仪进行pe150模式测序。
[0143]
五、数据分析
[0144]
使用trimmomatic去除接头以及低质量序列得到clean data,然后使用samtools提取98gene rna-cap panel目标区域的reads,统计目标基因的富集效率和覆盖参数。
[0145]
如表14所示,两个rna预文库(rna-重复1#和rna-重复2#)经98gene rna-cap panel杂交捕获的测序数据,取1000mb数据量进行分析,碱基质量q20均为98.2%,数据clean ratio为94.5%和94.7%,高通量测序质量表现正常;目标基因的数据占比(中靶率)为81.2%和81.3%,富集效率~2700倍(表14)。
[0146]
表14 rna样本捕获数据表现
[0147][0148]
在目标基因覆盖率和覆盖深度方面,1)覆盖率小于100%的基因数量为11个,占总基因数量的11.2%,约1/10的基因不完全覆盖,并不清楚是由于样本中不存在相应的剪接体还是探针不工作造成的;2)相对覆盖深度小于0.2的基因数量为43个,占总基因数量的43.9%,约2/5的基因覆盖深度较低,并不清楚是由于样本中上述基因表达较低还是探针不工作导致(表15)。
[0149]
对于目标基因的覆盖率,由于转录本存在不同的剪接(transcript variant),根
据数据库的转录本数据设计的探针,在具体的某个rna样本中可能捕获不到相应的转录本。因此,基因覆盖率低于100%,可能是由于mrna剪接差异造成,也有可能是探针不工作。
[0150]
对于目标基因的覆盖深度,由于基因的表达量不同,会存在覆盖深度较低的基因,目前无法判定这些基因的覆盖深度是由于低表达造成的,还是探针不工作导致的。
[0151]
前期研究发现利用不同的rna样本的基因剪接和表达差异并不能对目标基因的覆盖率和覆盖深度进行互补验证。例如,从tcga数据库中下载的多个细胞系样本的rna-seq数据,对上述98个基因的表达情况进行统计的结果是,低表达量基因即使在不同细胞系中也并未出现明显改善。
[0152]
发明人的研究结果表明,由于转录本的不同剪接方式以及表达量差异,仅仅通过液相基因芯片98gene rna-cap panel捕获rna样本的测序结果,无法判断某些目标基因的低覆盖率和低覆盖深度表现是否是探针性能问题所导致的。
[0153]
表15 rna样本捕获数据中基因覆盖表现
[0154]
[0155]
[0156][0157]
实施例2
[0158]
1、dna预文库构建:
[0159]
对dna样本构建预文库,两个重复(dna-重复1#、dna-重复2#),dna样本信息:na12878细胞系gdna(coriell)。
[0160]
1.1超声打断:
[0161]
取100ng na12878 gdna进行超声打断,打断大小为200bp(供应商:covaris;型号:m220)。
[0162]
1.2末端修复:
[0163]
如表16所示,向二链合成产物中加入end prep mix4(供应商:诺唯赞;货号:nd607-02),涡旋混匀后,短暂离心。将配制好的反应液放入pcr仪,按照表17所示程序运行
(热盖75℃)。
[0164]
表16
[0165]
组分体积μl100ngna12878gdna超声产物50endprepmix415总体积65
[0166]
表17
[0167][0168]
1.3接头连接:
[0169]
反应结束后,向上一步产物中加入表18示试剂(供应商:诺唯赞;货号:nd607-02),涡旋混匀后,短暂离心,在20℃条件下孵育30min。
[0170]
表18
[0171]
组分体积μl末端修复产物65illumina接头vazyme-s2.5rapidligationbuffer225rapiddnaligase5无酶无菌水2.5总体积100
[0172]
1.4接头连接产物纯化:
[0173]
反应结束后,向反应液中加入90μl vahts dna clean beads(供应商:诺唯赞;货号:n411-03)进行产物纯化,纯化操作同“实施例1之2.6步骤”所述。加入22μl low-te缓冲液洗脱,取20μl产物进入下一步反应。
[0174]
1.5 index pcr扩增
[0175]
向20μl接头连接产物中加入25ul vahts hifi amplification mix和5ul index primer(供应商:诺唯赞;货号:n411-03),按照表19程序进行pcr扩增。
[0176]
表19
[0177][0178]
1.6 dna预文库纯化:
[0179]
反应结束后,向反应液中加入45μl vahts dna clean beads(供应商:诺唯赞;货
号:n411-03)进行产物纯化,纯化操作同“实施例1之2.6步骤”所述。加入42μl无酶无菌水缓冲液洗脱,将40μl上清液转移至新的pcr管中。
[0180]
1.7 dna预文库质检:
[0181]
使用qubit荧光计3.0(thermofisher)测量文库浓度。使用agilent 2100测量文库片段长度,产物主带集中在320bp,无接头二聚。
[0182]
2、rna、dna预文库测序
[0183]
将rna预文库(实施例1之步骤2.9)以及本实施例中的dna预文库在illumina novaseq6000高通量测序仪上测序,分别获得全转录组测序数据(wts,rna样本)和全基因组测序数据(wgs,dna样本)。
[0184]
3、数据分析
[0185]
使用trimmomatic去除接头以及低质量序列得到clean data,使用refseq109数据库中的19475个基因,统计wts和wgs数据中的上述基因的覆盖率和覆盖深度情况(图2)。
[0186]
1)目标基因的覆盖率:在wts数据中,共有6252个基因的覆盖率小于100%(图2,a、b),占总基因数量的32.1%,在wgs数据中,共有695个基因的覆盖率小于100%,占总基因数量的3.6%(图3),两种测序数据的覆盖率小于100%的基因的交集为380个基因,占总基因数量的1.95%。
[0187]
2)目标基因的覆盖深度:在wts数据中,共有8418个基因的相对覆盖深度小于0.2(图2,c、d),占总基因数量的43.2%,在wgs数据中,共有418个基因的相对覆盖深度小于0.2,占总基因数量的2.1%,两种测序数据的相对覆盖深度小于0.2的基因的交集为329个基因,占总基因数量的1.69%(图3)。
[0188]
3)将wts和wgs数据中,目标基因的覆盖率均小于100%的380个基因与目标基因的相对覆盖深度均小于0.2的329个基因再取并集,共得到479个基因,占总基因数量的2.46%(图3),所述479个基因被定义为平行捕获的黑名单基因(表20)。这些基因即便通过平行捕获验证,也无法判定其是否正常工作。
[0189]
上述结果表明,基因组测序可以对转录组数据的基因覆盖率和覆盖深度参数进行有效补充,如果进一步应用在捕获测序中,可解决mrna剪接和表达差异的问题(图4)。对于近两万个人类基因而言(19475个基因),在理论上超过97.5%的基因可以通过rna和dna样本的平行捕获验证液相基因芯片的捕获性能。
[0190]
表20平行捕获的黑名单基因
[0191]
ratio为93.2%和93.5%,高通量测序质量表现正常;目标基因的数据占比(中靶率)为69.8%和69.9%,富集效率为~9800倍,证明98gene rna-cap gene panel仍然适用于基因组的富集。
[0197]
表21 98gene rna-cap gene panel捕获dna样本的数据表现
[0198][0199]
rna捕获数据中覆盖率小于100%的基因为11个,这些基因在dna捕获中覆盖率均为100%(表22),说明探针工作正常,而在rna捕获中覆盖不完全,可能是mrna剪接方式的原因。
[0200]
rna捕获数据中相对覆盖深度小于0.2的基因数量为43个,这些基因在dna捕获中的相对覆盖深度集中在0.8~1.2之间(表23),均一性优异,证明上述探针的捕获能力正常。
[0201]
上述结果表明,通过dna样本捕获,可以有效克服rna样本中存在的mrna剪接和表达差异干扰,有效评估液相基因芯片中的探针性能。
[0202]
表22平行捕获的目标基因覆盖率
[0203][0204][0205]
表23平行捕获的目标基因覆盖深度(相对覆盖深度)
[0206]
[0207][0208]
实施例4
[0209]
在经过dna样本捕获验证后,目标基因覆盖率和覆盖深度均达到质控要求(覆盖率100%,相对覆盖深度≥0.2)。随后,我们使用质控合格的98gene rna-cap gene panel对rna融合标准品(供应商:菁良;货号:gw-opsm005)进行杂交捕获测序。
[0210]
rna预文库构建和98gene rna-cap gene panel杂交捕获实验操作如分别按照实施例1所述,随后在illumina novaseq6000高通量测序仪上测序。
[0211]
使用trimmomatic去除接头以及低质量序列得到clean data,然后使用star进行序列比对,最后使用star-fusion进行融合基因分析。分析结果表明,对于6个已知阳性的融合基因,98gene rna-cap gene panel均可有效检出,证明开发的针对转录本序列设计的液相基因芯片的质控方法是可行的。
[0212]
表24 98gene rna-cap gene panel融合基因检测
[0213][0214]
[0215]
综上所述,由于rna样本中存在的mrna剪接和表达差异干扰,仅仅通过rna捕获,无法准确的测量液相基因芯片中探针的工作效率,其中就包括基因的覆盖率和覆盖深度。在比较了全转录组与全基因组的测序结果后,我们得到了通过基因组的捕获性能评估探针工作效率的启示。
[0216]
将针对转录本序列设计的98gene rna-cap gene panel与dna预文库进行杂交捕获,然后进行高通量测序,结果表明,在rna样本中覆盖不完全(覆盖率小于100%)和覆盖深度较低(相对覆盖深度小于0.2)的区域,在dna捕获数据中完全表现正常,证明98gene rna-cap gene panel中的探针工作有效。
[0217]
转录本序列设计,针对rna靶向测序的液相基因芯片在设计、合成完成后,需要评估其性能,本专利方法有效克服了mrna剪接和表达差异的干扰,能够准确评估液相基因芯片性能。
[0218]
尽管本发明已经参考示例性实施方案进行了描述,但应理解本发明不限于公开的示例性实施方案。在不背离本发明的范围或精神的情况下,可对本发明说明书的示例性实施方案做多种调整或变化。权利要求的范围应基于最宽的解释以涵盖所有修改和等同结构与功能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1