一种2,3,3,3-四氟丙烯的制备方法与流程
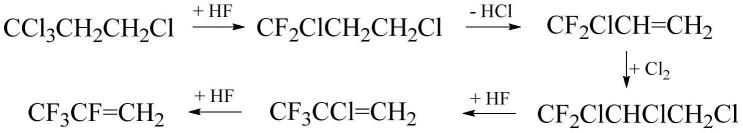
1.本发明公开了一种2,3,3,3-四氟丙烯的制备方法,尤其涉及以1,1,1,3-四氯丙烷为原料,经氟化、皂化、氯化及氟化反应制备2,3,3,3-四氟丙烯的方法。
背景技术:
2.2,3,3,3-四氟丙烯(hfo-1234yf),具有优异的理化和环境性能,成为有效的制冷剂、发泡剂、灭火剂、传热介质、推进剂、气态电介质、灭菌剂载体、动力循环工作流体、聚合物单体以及医药、农药中间体,应用广泛。
3.wo2009153493公开了一种以1,1,1,2,2,3-六氟丙烷(hfc-236cb)为原料,制备hfo-1234yf的方法,该方法首先是在氢气和催化剂ni-cr/alf3的存在下,hfc-236cb脱氟化氢生成1,2,3,3,3-五氟丙烯(hfo-1225ye),然后hfo-1225ye加氢得到1,1,1,2,3-五氟丙烷(hfc-245eb),最后再在氢气存在的条件下经脱氟化氢反应得到hfo-1234yf。
4.us20110190554公开了一种以1,1,2,3,3,3-六氟丙烯(hfp)为起始原料,经加氢、脱氟化氢、加氢、脱氟化氢四步反应合成hfo-1234yf的方法。
5.在以上合成方法中,原料成本较高,需要通入化学计量的氢气,且加氢步骤为了控制反应热需采用更高的摩尔比,而在较高的温度下通入过量的氢气会增加相关安全风险。
6.us2011207975公开了一种以1,1,2,3-四氯丙烯(tcp)或者1,1,1,2,3-五氯丙烷(hcc-240db)为原料合成hfo-1234yf的方法。该方法首先在cr2o3催化剂的存在下,在第一反应器中进行hf气相氟化tcp或者hcc-240db,得到2-氯-3,3,3-三氟丙烯(hcfc-1233xf),然后在sbcl5作用下,在第二反应器中液相氟化hcfc-1233xf得到2-氯-1,1,1,2-四氟丙烷(hcfc-244bb),最后在第三反应器中进行hcfc-244bb脱氯化氢反应得到hfo-1234yf。cn200980163128.7公开了一种由2,3-二氯-1,1,1-三氟丙烷(hcfc-243db)经hcfc-1233xf、hcfc-244bb制备hfo-1234yf的方法。在上述制备方法中,首先,使用的铬基催化剂会对人体造成损害,尤其是高价铬具有强致癌作用,可能会造成严重危害;其次,上述方法涉及到了沸点接近、具有类共沸的特性的中间体hcfc-1233xf和hcfc-244bb,而且这两种物质也均易与hf形成共沸物,三者分离工艺极其复杂,分离难度极大;最后,hcfc-244bb脱氯化氢制备hfo-1234yf时,含有的hcfo-1233xf及hf杂质能够严重影响脱氯化氢催化剂的寿命和产物选择性。
7.综上所述,目前制备hfo-1234yf的方法存在诸如原料不易得,催化剂对环境不友好,反应中间体难于分离,目标产物选择性低等不足,因此,需要持续改进,以获得更加有效的制备方法。
技术实现要素:
8.本发明的目的在于克服背景技术中存在的不足,提供一种原料易得、中间体易于分离、催化剂对环境友好的2,3,3,3-四氟丙烯的制备方法。
9.为了实现本发明的目的,本发明以1,1,1,3-四氯丙烷为原料,经液相氟化、皂化、
氯化、气相氟化反应制备2,3,3,3-四氟丙烯的方法。
[0010][0011]
为此,本发明提供的2,3,3,3-四氟丙烯的制备方法包括:
[0012]
(1)在氯磺酸催化作用下,烷烃为溶剂,及反应温度为0℃~100℃的条件下,1,1,1,3-四氯丙烷与三氟化锑反应制备1,3-二氯-1,1-二氟丙烷;
[0013]
(2)在相转移催化剂的作用下,及反应温度为60℃~150℃的条件下,1,3-二氯-1,1-二氟丙烷在碱性溶液中脱氯化氢制备3-氯-3,3-二氟丙烯;所述相转移催化剂为冠醚、季铵盐、季膦盐或聚乙二醇;所述碱性溶液为氢氧化钠的水溶液或氢氧化钾的水溶液;
[0014]
(3)在无催化剂或催化剂存在下,及反应温度为30℃~80℃的条件下,3-氯-3,3-二氟丙烯与氯气反应制备1,2,3-三氯-1,1-二氟丙烷;其中,催化剂为路易斯催化剂或离子盐催化剂;
[0015]
(4)在氟化催化剂fe-mo-mg-f的作用下,及反应温度为200℃~300℃的条件下,hf与1,2,3-三氯-1,1-二氟丙烷气相氟化制备2-氯-3,3,3-三氟丙烯;
[0016]
(5)在氟化催化剂a-b-al-f的作用下,及反应温度为250℃~350℃的条件下,hf与2-氯-3,3,3-三氟丙烯气相氟化制备2,3,3,3-四氟丙烯。
[0017]
可选的,步骤(1)中所述的烷烃为沸点大于150℃的烷烃,优选壬烷、癸烷或十二烷。
[0018]
可选的,步骤(1)中氯磺酸与1,1,1,3-四氯丙烷的摩尔比为0.1~1:1,三氟化锑与1,1,1,3-四氯丙烷摩尔比为0.5~0.8:1,反应时间为2h~10h。优选,所述的氯磺酸与1,1,1,3-四氯丙烷的摩尔比为0.3~0.5:1,三氟化锑与1,1,1,3-四氯丙烷摩尔比为0.6~0.7:1,反应时间为3h~5h。
[0019]
可选的,步骤(2)中所述的冠醚为环糊精;所述的季铵盐为四丁基氯化铵、四丁基溴化铵、苄基三乙基氯化铵或十二烷基三甲基氯化铵;所述的季膦盐为三苯基膦、四丁基溴化膦、十六烷基三丁基溴化膦、甲基三苯基溴化膦或四丁基醋酸膦;所述的聚乙二醇为聚乙二醇-4000、聚乙二醇-6000或聚乙二醇-8000。
[0020]
可选的,步骤(2)中所述相转移催化剂质量为1,3-二氯-1,1-二氟丙烷质量的0.5%~5%,碱与1,3-二氯-1,1-二氟丙烷的摩尔比为1~5:1,反应时间为1h~24h。优选,所述的相转移催化剂为1,3-二氯-1,1-二氟丙烷质量的1%~2%,碱性溶液为氢氧化钾的水溶液,碱性溶液质量浓度为40%~60%,碱与1,3-二氯-1,1-二氟丙烷的摩尔比为1.2~1.5:1,反应时间为5h~10h。
[0021]
可选的,步骤(3)中所述的催化剂为三氯化铁、三氯化铝或含氟钽盐催化剂,通式为q
+
[ta
x
cl
yf5x-y+1
]-,阳离子q+为季铵阳离子,1<x≤3,0≤y≤5。
[0022]
可选的,所述的季铵阳离子q
+
是四乙基铵、十二烷基三甲基铵、苯基三甲基铵、1-丁基-3-甲基咪唑鎓、1-丁基-2,3-二甲基咪唑鎓、n-丁基吡啶鎓、n-苯甲基吡啶鎓、n-丁基-n-甲基哌啶鎓或n-丁基-n-甲基吡咯烷鎓。
[0023]
可选的,步骤(3)中催化剂用量为3-氯-3,3-二氟丙烯质量的0.5%~10%,氯气与
3-氯-3,3-二氟丙烯摩尔比为1~10:1,反应时间为0.5h~10h。优选,所述的催化剂用量为3-氯-3,3-二氟丙烯质量的1%~3%,氯气与3-氯-3,3-二氟丙烯摩尔比为1.5~10:1,反应时间为2h~5h。
[0024]
可选的,步骤(4)中所述的氟化催化剂fe-mo-mg-f,fe、mo与mg三者的摩尔比为(2~4)∶(0.5~1)∶(5~7.5)。
[0025]
可选的,步骤(4)hf与1,2,3-三氯-1,1-二氟丙烷的摩尔比为3~100:1,接触时间1~30s。优选,步骤(4)中所述的反应温度为240℃~280℃,hf与1,2,3-三氯-1,1-二氟丙烷的摩尔比为10~30:1,接触时间10s~20s。
[0026]
可选的,步骤(5)中所述的氟化催化剂a-b-al-f,a为第
ⅷ
族元素,优选ni、fe或co中的一种,b为高场强元素,优选sc、y、th、u、pb以及zr中的一种。
[0027]
可选的,步骤(5)中所述的氟化催化剂a-b-al-f,a、b与al三者的摩尔比为(3~4)∶(0.1~0.5)∶(5.5~6.9)。
[0028]
可选的,步骤(5)中hf与2-氯-3,3,3-三氟丙烯的摩尔比为2~100:1,接触时间1~20s。优选,步骤(5)中所述的反应温度为300℃~320℃,hf与2-氯-3,3,3-三氟丙烯的摩尔比为10~20:1,接触时间10s~15s。
[0029]
与现有技术相比,本发明的有益效果如下:本发明提供了一种有效合成hfo-1234yf的方法,原料1,1,1,3-四氯丙烷易得;反应催化剂不涉及含铬、含锑化合物,对人和环境友好;反应条件温和,不涉及到氢气;产物选择性较高:第一步1,3-二氯-1,1-二氟丙烷选择性达到95.3%,第二步,3-氯-3,3-二氟丙烯选择性达到99.8%,第三步1,2,3-三氯-1,1-二氟丙烷选择性达到97.5%,第四步2-氯-3,3,3-三氟丙烯选择性达到99.0%,第五步2,3,3,3-四氟丙烯选择性达到88.5%。
附图说明
[0030]
图1为本发明实施例1制备的1,3-二氯-1,1-二氟丙烷质谱结果。
[0031]
图2为本发明实施例12制备的3-氯-3,3-二氟丙烯质谱结果。
[0032]
图3为本发明实施例32制备的1,2,3-三氯-1,1-二氟丙烷质谱结果。
[0033]
图4为本发明实施例50制备的2-氯-3,3,3-三氟丙烯质谱结果。
具体实施方式
[0034]
除非有特殊说明,本文中的科学与技术术语根据相关领域普通技术人员的认识理解。还应理解,本文涉及的温度、浓度是近似值,用于说明目的。虽然与本文描述的方法和材料相似或等价的方法和材料可以用于本公开的实施,但下文描述了部分适合的方法和材料。本文提到的出版物、专利申请、专利和其他参考文献以引用方式部分纳入本文,如出现冲突,以本文为准。另外,所述材料、方法、溶液浓度和实施例仅是示例性的,而并不意欲进行限制。具体方案中,本领技术人员可以根据本发明所公开内容采用常规实验时段对方法中所涉及的物质配比、浓度、操作参数取值进行优化以实现本发明的目的。
[0035]
本发明中,反应的操作压力主要由反应温度下反应物的饱和蒸气压所控制,一般不作严格控制,可在低于、等于或高于大气压下进行,优选在高于大气压下进行。此外,本发明反应可以间歇操作,也可连续操作,反应本身对反应形式无明显要求。
[0036]
本发明中三氟化锑在使用前进行脱水处理,优选进行升华处理。
[0037]
本发明所述的含氟钽盐催化剂中,阴离子[ta
x
cl
yf5x-y+1
]-为催化中心,x和y值合适时,含氟钽盐具有优良的催化活性和稳定性,x和y值较优的范围分别为1<x≤2.5,0≤y≤3,更优选的范围分别为1.5<x≤2,0≤y≤1。
[0038]
所述的含氟钽盐制备方法如下:在溶剂中将氟化氢盐q
+
f-(hf)m(0<m<20)与钽盐tacl
nf5-n
(n=0~5),以1:1~3的摩尔比例混合反应,温度为25~100℃,时间为1~24h,随后脱除溶剂得到含氟钽盐q
+
[ta
x
cl
yf5x-y+1
]-。方法中所述的溶剂为不与路易斯酸和氟化氢发生化学反应的极性溶剂,具体优选的有so2、soclf、ch3cn、cf3ch2cf2、cf3ch2cf2ch3等。
[0039]
本发明步骤(4)中所述的氟化催化剂fe-mo-mg-f,的制备方法有浸渍法、共沉淀法、共混法、溶胶-凝胶法等催化剂制备方法。该催化剂的预处理可以通过将催化剂在氮气或其他惰性气体流中加热到约200℃至约400℃来进行。然后催化剂可以用经大量过量的氮气稀释的氟化氢流进行活化以得到高催化剂活性。催化剂的再生可以在如下条件下进行,使空气或用氮气稀释的空气在约100℃至约380℃,优选约150℃至约350℃的温度处理催化剂,持续约8小时至约48小时。
[0040]
本发明步骤(5)所述的气相氟化催化剂制备方法包括:(1)搅拌下,将氢氟酸滴入磷酸铝的1,2-丙二醇溶液中进行氟化处理,氢氟酸与磷酸铝摩尔比为3~6:1,滴加完毕后继续搅拌8h~10h以上得到液体溶胶;将液体溶胶在70℃~80℃静置老化24h~48h得到固体凝胶;将固体凝胶在120℃~140℃干燥30h,得到高比表面积的alf3活性载体;(2)通过共浸渍的方法,将活性组分a和助剂b负载在alf3载体上,于100℃~120℃干燥,最后在280℃~350℃下焙烧,得到所需的a-b-al-f复合催化剂。
[0041]
下列结合实施例对本发明进一步详述说明,但并不限制本发明的范围。以下实施例中反应物的转化率及选择性检测采用gc-ms检测方法。
[0042]
实施例1
[0043]
氟化制备1,3-二氯-1,1-二氟丙烷。
[0044]
向300ml不锈钢带搅拌高压反应釜中,依次加入16.7g氯磺酸、35.6g三氟化锑和50ml的壬烷以及52g 1,1,1,3-四氯丙烷,开启搅拌,升温至60℃后反应5h,反应结束后取样经水洗、过滤、除酸后用气相色谱分析,1,1,1,3-四氯丙烷的转化率为99.7%,1,3-二氯-1,1-二氟丙烷选择性为95.3%。
[0045]
1,3-二氯-1,1-二氟丙烷的gc-ms结果如图1所示,m/z113为cclf2ch2ch2cl脱cl后离子峰,m/z93为cclf2ch2ch2cl脱cl和f后离子峰,m/z85为cclf2离子峰,m/z77为cclf2ch2ch2cl脱2个cl后离子峰,m/z51为二氟甲基。上述数据证明制得产物就是1,3-二氯-1,1-二氟丙烷。
[0046]
实施例2~7:
[0047]
实施例2~7按照实施例1中相同的方法制备1,3-二氯-1,1-二氟丙烷,所不同的是,在不改变各反应物配比的前提下,改变溶剂、反应温度和反应时间,反应结果如表1所示。
[0048]
表1
[0049][0050]
实施例7~11:
[0051]
实施例7~11按照实施例1中相同的方法制备1,3-二氯-1,1-二氟丙烷,所不同的是三氟化锑与1,1,1,3-四氯丙烷的摩尔比、催化剂与1,1,1,3-四氯丙烷的摩尔比,反应结果如表2所示。
[0052]
表2
[0053][0054]
实施例12:
[0055]
碱脱制备3-氯-3,3-二氟丙烯。
[0056]
在带搅拌的250ml不锈钢高压釜中进行。向反应釜中依次投入100g 50%的koh水溶液,60g 1,3-二氯-1,1-二氟丙烷和1wt%环糊精(6g),开启搅拌,将反应温度控制在100℃,反应10h后,反应物料冷却至室温,相分后收集有机物进行气相色谱分析,结果表明1,3-二氯-1,1-二氟丙烷的转化率为99.0%,3-氯-3,3-二氟丙烯的选择性为99.8%。
[0057]
3-氯-3,3-二氟丙烯的gc-ms结果如图2所示,存在m/z113为分子离子峰,m/z93为cclf2ch=ch2脱f后离子峰,m/z85为二氟氯甲基,m/z77为cclf2ch=ch2脱氯后离子峰,m/z51为二氟甲基。上述数据证明制得产物就是3-氯-3,3-二氟丙烯。
[0058]
实施例13~24:
[0059]
实施例13~24按照实施例12中相同的方法制备3-氯-3,3-二氟丙烯,所不同的是改变相转移催化剂种类及含量、反应温度和反应时间,反应结果如表3所示。
[0060]
表3
[0061][0062]
实施例25:
[0063]
实施例25按照实施例12中相同的方法制备3-氯-3,3-二氟丙烯,所不同的是改变碱液浓度为naoh水溶液,结果表明1,3-二氯-1,1-二氟丙烷的转化率为98.5%,3-氯-3,3-二氟丙烯的选择性为97.8%。
[0064]
实施例26~31:
[0065]
实施例26~31按照实施例12中相同的方法制备3-氯-3,3-二氟丙烯,所不同的是改变碱液浓度、碱与1,3-二氯-1,1-二氟丙烷的摩尔比,反应结果如表4所示。
[0066]
表4
[0067][0068][0069]
实施例32:
[0070]
制备1,2,3-三氯-1,1-二氟丙烷。
[0071]
向250ml不锈钢带搅拌高压反应釜中加入56.3g 3-氯-3,3-二氟丙烯,5.63g[net4]ta2cl5f6,升温至反应温度40℃,开启搅拌后通入氯气53.3g,维持反应压力0.3mpa反应5h,降温停止反应,取样进行gc分析,3-氯-3,3-二氟丙烯转化率为99.8%,1,2,3-三氯-1,1-二氟丙烷选择性为97.5%。
[0072]
1,2,3-三氯-1,1-二氟丙烷的gc-ms结果如图3所示,存在m/z182为分子离子峰,m/z147为cf2clchclch2cl脱cl后离子峰,m/z133为cf2clchclch2cl脱ch2cl后离子峰,m/z111为cf2clchclch2cl脱两个cl后离子峰,m/z97为cf2clchclch2cl脱cf2cl后离子峰,m/z85为cf2cl,m/z49为ch2cl。上述数据证明制得产物就是1,2,3-三氯-1,1-二氟丙烷。
[0073]
实施例33~44:
[0074]
实施例33~44按照实施例32中相同的方法制备1,2,3-三氯-1,1-二氟丙烷,所不同的是催化剂与其用量,反应结果如表5所示。
[0075]
表5
[0076][0077][0078]
实施例45~49:
[0079]
实施例45~49按照实施例32中相同的方法制备1,2,3-三氯-1,1-二氟丙烷,所不同的是氯气与3-氯-3,3-二氟丙烯摩尔比、反应温度及反应时间,反应结果如表6所示。
[0080]
表6
[0081][0082]
实施例50:
[0083]
气相氟化反应合成2-氯-3,3,3-三氟丙烯。
[0084]
催化剂的制备:按一定比例将mo2o3加到mg(no3)2·
6h2o和fe(no3)2·
9h2o的混合水溶液中,加质量分数为12%的碳酸氢氨,调节ph控制为8~9,约5h后经洗涤后离心分离,再在120℃下烘干,再在200℃下焙烧1h,5℃/min升至300℃,焙烧2h,然后5℃/min升至400℃,焙烧4h,最后再依次通过氟化氢活化处理得到fe-mo-mg-f催化剂。
[0085]
在内径为38mm的固定床管式反应器中,装入50ml上述fe-mo-mg-f催化剂,其中,fe、mo、mg三者的摩尔比为3.4:0.6:6,待反应温度稳定在280℃后,分别通入hf和1,2,3-三氯-1,1-二氟丙烷,在常压下进行反应,控制两者的摩尔比为30:1,接触时间为20s,,运行24h,反应产物经水洗hf、-10℃低温收集并干燥后,气相色谱进行分析,结果表明1,2,3-三氯-1,1-二氟丙烷的转化率为100%,2-氯-3,3,3-三氟丙烯的选择性为99.1%。
[0086]
2-氯-3,3,3-三氟丙烯的gc-ms结果如图4所示,存在m/z130为分子离子峰,m/z111为cf3ccl=ch2脱f后离子峰,m/z95为cf3ccl=ch2脱cl后离子峰,m/z69为cf3离子峰,m/z61为ccl=ch2离子峰。上述数据证明制得产物就是2-氯-3,3,3-三氟丙烯。
[0087]
通过将上述反应液精馏收集沸点为13℃-14℃的产品,纯度99.5%,经1h-nmr、
13
cnmr、
19
f-nmr表征鉴定为2-氯-3,3,3-三氟丙烯。
[0088]1h-nmr(cdcl3)δ5.195(s,1h),5.101(s,1h),j
h-f
=47hz;
[0089]
13
c-nmr(cdcl3)δ127.112-127.246(d,1c,j
c-f
=67hz),124.724-124.811(d,1c,j
c-f
=43.5hz),80.343-80.733(d,1c,j
c-f
=195hz);
[0090]
19
f-nmr(cdcl3)δ-68.066(s,3f)。
[0091]
实施例51~54:
[0092]
实施例51~54气相氟化反应合成2-氯-3,3,3-三氟丙烯与实施例50相同,所不同的是催化剂组分,反应结果如表7所示。
[0093]
表7
[0094][0095]
实施例55~59:
[0096]
实施例55~59按照实施例50中相同的方法制备2-氯-3,3,3-三氟丙烯,所不同的是hf与1,2,3-三氯-1,1-二氟丙烷的摩尔比、反应温度及接触时间,反应结果如表8所示。
[0097]
表8
[0098][0099][0100]
实施例60:
[0101]
气相氟化反应合成2,3,3,3-四氟丙烯。
[0102]
催化剂的制备:搅拌下,将氢氟酸滴入磷酸铝的1,2-丙二醇溶液中进行氟化处理,氢氟酸与磷酸铝摩尔比为6:1,滴加完毕后继续搅拌~10h得到液体溶胶;将液体溶胶在70℃~80℃静置老化48h得到固体凝胶;将固体凝胶在120℃干燥30h,得到高比表面积的alf3活性载体;通过共浸渍的方法,将活性组分co和助剂y负载在alf3载体上,于120℃干燥,最后在350℃下焙烧,得到co-y-al-f复合催化剂。
[0103]
在内径为38mm的固定床管式反应器中,装入50ml含上述co-y-al-f催化剂,其中,co、y、al三者的摩尔比为3.5:0.3:6.2,待反应温度稳定在320℃后,分别通入hf和2-氯-3,3,3-三氟丙烯,在常压下进行反应,控制两者的摩尔比为30:1,接触时间为10s,运行24h,反应产物经水洗hf、-40℃低温带压钢瓶收集,气相色谱进行分析,结果表明2-氯-3,3,3-三氟丙烯的转化率为82.6%,2,3,3,3-四氟丙烯的选择性为88.7%。
[0104]
通过将上述反应液精馏收集沸点为-28℃的产品,纯度99.5%,经1h-nmr、
13
cnmr、
19
f-nmr表征鉴定为2,3,3,3-四氟丙烯。
[0105]1h-nmr(cdcl3)δ5.195(s,1h),5.101(s,1h),j
h-f
=47hz;
[0106]
13
c-nmr(cdcl3)δ127.112-127.246(d,1c,j
c-f
=67hz),124.724-124.811(d,1c,j
c-f
=43.5hz),80.343-80.733(d,1c,j
c-f
=195hz);
[0107]
19
f-nmr(cdcl3)δ-68.066(s,3f)。
[0108]
实施例61~64:
[0109]
实施例61~64气相氟化反应合成2,3,3,3-四氟丙烯与实施例60相同,所不同的是催化剂种类和组分,反应结果如表9所示。
[0110]
表9
[0111][0112][0113]
实施例67~71:
[0114]
实施例67~71按照实施例60中相同的方法制备2,3,3,3-四氟丙烯,所不同的是hf与2-氯-3,3,3-三氟丙烯的摩尔比、反应温度及接触时间,反应结果如表10所示。
[0115]
表10
[0116][0117]
以上所述,仅是本发明的部分实施例而已,并非对本发明做任何形式上的限制,凡是依据本发明的技术实质对上述实施例作的任何简单的修改,等同变化与修饰,均属于本发明技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1