N的制作方法
n
ε-十二酰基赖氨酸的制备方法
技术领域
1.本发明涉及n
ε-十二酰基赖氨酸的制备方法,属于化工合成技术领域。
背景技术:
2.n
ε-十二酰基赖氨酸属于氨基酸类衍生物,具备天然生物原料带给它的高安全性、能够生物降解等特点,使其在日化、食品等行业得到广泛的关注和应用。n
ε-十二酰基赖氨酸是一种对溶剂不敏感的疏水粉体,片状结晶结构赋予它优异的润滑性,是一种理想的化妆品基质。
3.中国专利cn107417561a和中国专利cn107488130a报道:以月桂酸和l-赖氨酸为原料,甲醇为溶剂,回流反应,降温过滤得赖氨酸月桂酸盐;再将赖氨酸月桂酸盐脱水,降温过滤得月桂酰赖氨酸粗品;粗品经过重结晶后得月桂酰赖氨酸。该方法工序步骤较多,操作过程较为复杂,收率分别在50%和35%左右。
4.中国专利cn102617390a报道:以赖氨酸或其盐、十二酰氯为原料,首先使赖氨酸或其盐与二价金属离子形成螯合物,然后该螯合物与月桂酰氯进行酰胺化反应,最后通过酸解破坏螯合结构得n-月桂酰赖氨酸。该方法虽然副产物少,但废水量大,收率不足50%。
技术实现要素:
5.本发明针对现有技术存在的不足,提供一种n
ε-十二酰基赖氨酸的制备方法,所述制备方法在催化剂的作用下提高两分子间赖氨酸α-氨基和α-羧基环合的选择性,通过形成肽键实现α位基团的保护来降低副反应,大大提高目标产品的收率。
6.本发明解决上述技术问题的技术方案如下:一种n
ε-十二酰基赖氨酸的制备方法,所述的制备方法包括如下步骤:s1、环合反应:在有机溶剂中加入l-赖氨酸和催化剂,加热进行环合反应制得ε-氨基-单环-α-丁酰胺环合反应液;所述催化剂为dmap(4-二甲氨基吡啶)、dmapo(4-二甲胺基吡啶n-氧化物)和4-ppy(4-吡咯烷基吡啶)中的一种或者多种组合;s2、酰胺化反应:在一定温度下,向步骤s1的ε-氨基-单环-α-丁酰胺环合反应液中滴加十二酰氯,并加入缚酸剂调节体系ph值,经过酰胺化反应得到酰胺化反应液;s3、水解反应:将步骤s2的酰胺化反应液用缚酸剂配制的水溶液调节体系ph,在一定温度下经过水解反应开环,反应毕,盐酸调节ph至中性,制得n
ε-十二酰基赖氨酸。
7.进一步的,步骤s1中,所述的催化剂为dmap、dmap与dmapo的组合、dmap与4-ppy的组合或者dmap、dmapo与4-ppy的组合。
8.进一步的,步骤s1中,所述的有机溶剂为n,n-二甲基乙酰胺、n-甲基-2-吡咯烷酮、甲苯、对二甲苯、间二甲苯、邻二甲苯和三甲苯中的一种或者两种的组合。
9.优选的,步骤s1中,所述的有机溶剂为高沸点溶剂与低沸点溶剂的组合,所述高沸点溶剂与低沸点溶剂的沸点差不小于25℃,所述低沸点溶剂的沸点不小于100℃,所述的低沸点溶剂为与甲苯、对二甲苯、间二甲苯、邻二甲苯任意一种,所述的高沸点溶剂为n-甲基-2-吡咯烷酮。
10.优选的,所述高沸点溶剂与低沸点溶剂的质量比为3~5:1,步骤s2的酰胺化反应结束后蒸出低沸点溶剂再进行步骤s3的水解反应。
11.优选的,步骤s1中,所述的有机溶剂为n-甲基-2-吡咯烷酮、甲苯、二甲苯和三甲苯中的一种或者两种的组合。
12.进一步的,步骤s1中,所述催化剂与l-赖氨酸的质量比为0.001~0.3:1,所述有机溶剂与l-赖氨酸的质量比为3~15:1,l-赖氨酸与十二酰氯的摩尔比为1:1~2.5。
13.优选的,步骤s1中,所述催化剂与l-赖氨酸的质量比为0.05-0.1:1;所述有机溶剂与l-赖氨酸的质量比为3-12:1;l-赖氨酸与十二酰氯的摩尔比为1:1~1.5。
14.进一步的,步骤s1中,环合反应温度为110~200℃。
15.优选的,步骤s1中,环合反应温度为110~170℃。
16.进一步的,步骤s2中,酰胺化反应温度为-25~15℃。
17.优选的,步骤s2中,酰胺化反应温度为-10~15℃。
18.进一步的,步骤s2中,加入缚酸剂调节体系ph值为7~10。
19.优选的,步骤s2中,加入缚酸剂调节体系ph值为7~8。
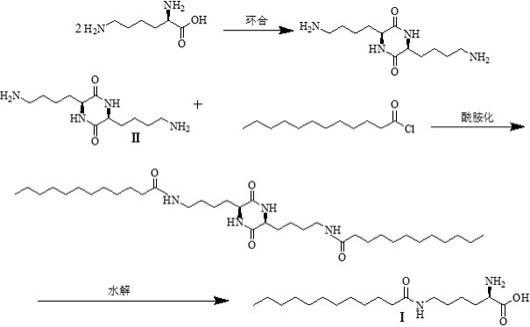
20.进一步的,步骤s2和步骤s3中,所述的缚酸剂为koh、khco3、k2co3和naoh中的一种或者多种组合。
21.优选的,步骤s2和步骤s3中,所述的缚酸剂为koh。
22.进一步的,步骤s3中,酰胺化反应液用缚酸剂配制的水溶液调节体系ph为7~13。
23.优选的,步骤s3中,酰胺化反应液用缚酸剂配制的水溶液调节体系ph为9~13。
24.进一步的,步骤s3中,水解反应温度为40~100℃。
25.优选的,步骤s3中,水解反应温度为40~80℃。
26.本发明的有益效果是:(1)本发明所述制备方法中,通过环合反应形成肽键实现对α位基团的保护来降低副反应,大大提高目标产品的收率,产品纯度和收率较高。
27.(2)环合反应中的催化剂有效提高了两分子赖氨酸间的α-氨基和α-羧基环合的选择性,从而更利于得到收率高的产品。
28.(3)酰胺化反应中,通过高沸点溶剂与低沸点溶剂的配合使用,更利于反应过程的进行,酰胺化反应结束后蒸出低沸点溶剂可以确保反应体系中的水分被充分蒸出,利于反应进程充分,减少体系内杂质,从而提高最终产品收率和纯度。
29.(4)本发明所述制备方法中,通过控制酰胺化反应温度和酰胺化反应体系的ph值,控制反应过程,得到高收率的n
ε-十二酰基赖氨酸产品。
30.(5)本发明所述制备方法操作安全简单,原料成本较低,符合绿色环保的生产理念,所述制备方法克服了现有技术存在的高成本、低收率等难题,有利于工业化生产。
附图说明
31.图1为本发明所述n
ε-十二酰基赖氨酸制备方法的反应路线;图2为实施例1制备的n
ε-十二酰基赖氨酸的红外谱图;图3为重水条件下的实施例1制备的n
ε-十二酰基赖氨酸的氢核磁谱图。
具体实施方式
32.下面对本发明的具体实施方式做详细说明。本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受公开的具体实施例的限制。
33.除非另有定义,本文所使用的所有技术和科学术语与本发明所属技术领域的技术人员通常理解的含义相同。所使用的术语只为描述具体实施方式,不为限制本发明。
34.实施例1向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮104g和对二甲苯26g,升温至140℃回流分水,反应2h,蒸除对二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
35.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸31.6g。纯度99.84%,总收率96.3%。红外谱图和氢核磁谱图分别见图2和图3。
36.实施例2向烧瓶投入l-赖氨酸14.6g、dmap 1.5g、n-甲基-2-吡咯烷酮120g和三甲苯30g,升温至160℃回流分水,反应2h,蒸除三甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
37.上述反应液在0℃滴加十二酰氯24g,koh调节ph值至7,保温1h得酰胺化反应液。酰胺化反应液用koh水溶液调节体系ph至11,并升温至70℃反应5h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 31g。纯度99.79%,总收率94.5%。
38.实施例3向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮104g和邻二甲苯26g,升温至120℃回流分水,反应5h,蒸除邻二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
39.上述反应液在10℃滴加十二酰氯23g,naoh调节ph值至7,保温1h得酰胺化反应液。酰胺化反应液用naoh水溶液调节体系ph至10,并升温至75℃反应5h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 30g。纯度99.75%,总收率91.5%。
40.实施例4向烧瓶投入l-赖氨酸14.6g、dmapo 0.7g、n-甲基-2-吡咯烷酮104g和间二甲苯26g,升温至140℃回流分水,反应2h,蒸除间二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
41.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 29.5g。纯度99.79%,总收率90%。
42.实施例5
向烧瓶投入l-赖氨酸14.6g、4-ppy 1.2g、n-甲基-2-吡咯烷酮104g和对二甲苯26g,升温至140℃回流分水,反应2h,蒸除对二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
43.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 25.6g。纯度99.70%,总收率78%。
44.实施例6向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮104g和邻二甲苯26g,升温至140℃回流分水,反应2h,蒸除邻二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
45.上述反应液在-5℃滴加十二酰氯22.9g,khco3调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用khco3水溶液调节体系ph至10,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 24.6g。纯度99.65%,总收率75%。
46.实施例7向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮104g和间二甲苯26g,升温至140℃回流分水,反应2h,蒸除二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
47.上述反应液在-5℃滴加十二酰氯22.9g,k2co3调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用k2co3水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸26.7g。纯度99.72%,总收率82%。
48.实施例8向烧瓶投入l-赖氨酸14.6g、dmapo 3.65g、n,n-二甲基乙酰胺216g,升温至195℃回流分水,反应2h,得ε-氨基-单环-α-丁酰胺化反应液。
49.上述反应液在-15℃滴加十二酰氯32.8g,koh调节ph值至7,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至9,并升温至40℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸 28.11g。纯度99.76%,总收率85.7%。
50.实施例9向烧瓶投入l-赖氨酸14.6g、dmap 0.15g、三甲苯44g,升温至170℃回流分水,反应2h,得ε-氨基-单环-α-丁酰胺化反应液。
51.上述反应液在-25℃滴加十二酰氯54.5g,naoh调节ph值至10,保温1h得酰胺化反应液。随后酰胺化反应液用naoh水溶液调节体系ph至13,并升温至95℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸29.6g。纯度99.72%,总收率90.2%。
52.实施例10向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮130g,升温至140℃回流分水,反应2h,得ε-氨基-单环-α-丁酰胺化反应液。
53.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%
盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸29.4g。纯度99.84%,总收率89.50%。
54.实施例11向烧瓶投入l-赖氨酸14.6g、dmap 0.4g、dmapo 0.3g、n-甲基-2-吡咯烷酮104g和间二甲苯26g,升温至140℃回流分水,反应2h,蒸除间二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
55.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸31.7g。纯度99.79%,总收率96.5%。
56.实施例12向烧瓶投入l-赖氨酸14.6g、dmap 0.6g、4-ppy 0.6g、n-甲基-2-吡咯烷酮104g和对二甲苯26g,升温至140℃回流分水,反应2h,蒸除对二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
57.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸31.62g。纯度99.70%,总收率96.4%。
58.实施例13向烧瓶投入l-赖氨酸14.6g、dmap 0.8g、dmapo 0.2g、4-ppy 0.2g、n-甲基-2-吡咯烷酮104g和对二甲苯26g,升温至140℃回流分水,反应2h,蒸除对二甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
59.上述反应液在-5℃滴加十二酰氯22.9g,koh调节ph值至8,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至12,并升温至80℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸31.7g。纯度99.84%,总收率96.5%。
60.实施例14向烧瓶投入l-赖氨酸14.6g、dmap 1.2g、n-甲基-2-吡咯烷酮60g和甲苯20g,升温至150℃回流分水,反应2h,蒸除甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
61.上述反应液在-15℃滴加十二酰氯25.6g,koh调节ph值至9,保温1h得酰胺化反应液。随后酰胺化反应液用koh水溶液调节体系ph至13,并升温至75℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸31.4g。纯度99.80%,总收率95.7%。
62.实施例15向烧瓶投入l-赖氨酸14.6g、dmap 1.5g、n-甲基-2-吡咯烷酮150g和三甲苯30g,升温至130℃回流分水,反应2h,蒸除三甲苯后得ε-氨基-单环-α-丁酰胺化反应液。
63.上述反应液在15℃滴加十二酰氯24g,naoh调节ph值至9,保温1h得酰胺化反应液。随后酰胺化反应液用naoh水溶液调节体系ph至11,并升温至98℃反应4h,反应毕,用30%盐酸溶液调节ph至中性,产品析出、过滤、烘干得n
ε-十二酰基赖氨酸30.3g。纯度99.79%,总收
率92.5%。
64.对比例1采用实施例1相同的方法制备n
ε-十二酰基赖氨酸,不同之处在于:酰胺化反应温度为20℃。最终得n
ε-十二酰基赖氨酸20.5g。纯度99.72%,总收率62.5%。
65.对比例2采用实施例1相同的方法制备n
ε-十二酰基赖氨酸,不同之处在于:酰胺化反应的ph为11。最终得n
ε-十二酰基赖氨酸19.8g。纯度99.79%,总收率60.3%。
66.对比例3采用实施例1相同的方法制备n
ε-十二酰基赖氨酸,不同之处在于:环化反应温度为100℃。最终得n
ε-十二酰基赖氨酸19.8g。纯度99.82%,总收率61.4%。
67.对比例4在50℃下,向反应瓶加入10gl-赖氨酸、300ml甲醇,然后加入16.5g十二酰氯,用氢氧化钠调节混合液的ph值至10,搅拌反应2h,反应停止后,用盐酸将反应液的ph值调至8,过滤,将过滤物用100ml甲醇分批次洗涤,再次过滤,将滤物用乙醇水溶液洗涤过滤,干燥得高纯度n
ε-十二酰基赖氨酸产品11.41g。纯度99.65%,总收率50.8%。
68.实施例1-15和对比例1-4的工艺条件及结果如下表1-表4所示:表1实施例1-6的工艺条件及结果数据
表2实施例7-12的工艺条件及结果数据
表3实施例13-15的工艺条件及结果数据
表4对比例1-4的工艺条件及结果数据
从实施例1-15的实验数据可以看出,采用本发明所述制备方法制备n
ε-十二酰基赖氨酸,产品收率能够达到75%以上,甚至能够达到95%以上,说明本发明所述制备方法中通过环合反应形成肽键实现对α位基团的保护,可以有效降低副反应,大大提高目标产品的收率,产品纯度和收率较高。
69.对比例4中采用目前公开的常规方法制备n
ε-十二酰基赖氨酸,产品收率只有50.8%,明显低于本发明所述制备方法的收率。
70.通过实施例1和对比例1、对比例2的数据比对可以看出,采用本发明所述酰胺化反应温度和酰胺化反应ph的条件,更利于得到收率较高的n
ε-十二酰基赖氨酸产品。
71.通过实施例1和对比例3的数据比对可以看出,采用本发明所述环合反应温度条件,更利于得到收率较高的n
ε-十二酰基赖氨酸产品。
72.另外,通过实施例1和实施例13的数据比对可以看出,采用高沸点溶剂和低沸点溶剂的组合更利于得到高收率的n
ε-十二酰基赖氨酸产品,采用高沸点溶剂和低沸点溶剂的组合进行环合反应后,将低沸点溶剂蒸出再进行酰胺化反应,可以确保环合反应产生的水分被完全脱除,从而更利于n
ε-十二酰基赖氨酸产品的制备。
73.通过实施例4和实施例10的数据比对;实施例5和实施例11的数据比对;实施例1、实施例4、实施例5和实施例12的数据比对可以看出,催化剂采用dmap和dmapo的组合、dmap和4-ppy的组合、dmap与dmapo和4-ppy的组合,更利于得到高收率的n
ε-十二酰基赖氨酸产品。
74.以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合穷举,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
75.对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围,本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1