聚集诱导发光材料的制备及在光动力治疗和生物成像中的应用
1.本发明属于聚集诱导发光材料领域,特别涉及聚集诱导发光材料的制备及在光动力治疗和生物成像中的应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.荧光成像技术凭借其快速的响应、卓越的时间分辨率、高超的灵敏度、原位可操作和良好的重现性,成为一种强大的、非侵入性的生物物种可视化分析工具。小型有机荧光团作为荧光材料的一个主要分支,目前正经历着爆炸性的发展,特别是具有近红外发射(》700nm)的荧光团,具有穿透深度高、生物自身荧光干扰低、光对生物结构的损伤最小、光散射减少等独特优势。
4.然而,由于π-π叠加和其他非辐射途径,传统的近红外荧光团是高浓度或聚集态时弱发射或非发射,这种现象被称为聚集引起的猝灭(acq)。这种现象非常常见,并且仍然是实现其在生物成像和治疗领域的实际应用的主要障碍,因为有机分子由于其发射中心的高疏水性,在生物介质中自然聚集。但是,一类具有聚集诱导发射(aie)特性的新型近红外荧光团的出现,完美地解决了acq问题。aie发光体(aiegens)在分子溶解于溶剂时是非发射的,但在聚集体中被诱导产生强烈的荧光。aie的特性允许使用任何浓度的荧光团溶液,并使开发用于生物传感和成像应用的荧光发光探针成为可能。目前近红外alegens的发展还远不够理想,到目前为止,只有少数表现出高性能近红外发射的alegens被开发并用于生物研究。
5.目前报道的alegens在结构上主要是含有苯环和含氧、含硫、含氮和含磷原子的杂环,但是含有硒原子的杂环至今尚未有报道。
6.同时,虽然有部分的alegens能够对某些特定的癌细胞有杀伤作用,但普遍存在杀伤率较低的问题,目前对于a549细胞具有较高的杀伤率的聚集诱导发光材料尚未见报道。
技术实现要素:
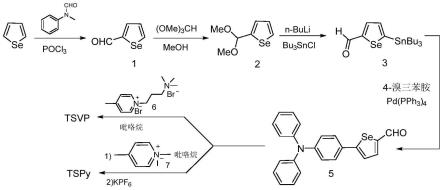
7.为了解决上述问题,本发明提供聚集诱导发光材料的制备及在光动力治疗和生物成像中的应用。本发明应用有机合成方法从商业可得的硒酚出发经过甲酰化、保护、锡化、金属催化的偶联、缩合等步骤得到含硒酚环的聚集诱导发光材料tsvp和tspy。
8.为了实现上述目的,本发明采用如下技术方案:
9.本发明的第一个方面,提供了两种聚集诱导发光材料,包括:所述聚集诱导发光材料为tsvp或tspy;
10.其中,tsvp的结构式如下:
[0011][0012]
其中,tspy的结构式如下:
[0013][0014]
本发明的第二个方面,提供了两种聚集诱导发光材料的合成方法,包括:
[0015]
将商业化的硒酚与三氯氧磷和n-甲基n-苯基甲酰胺混合均匀,室温搅拌反应,得到粗产品,后处理后脱除溶剂,纯化得到2-甲酰硒酚1;
[0016]
将2-甲酰硒酚1、甲醇、无水氯化铵、原甲酸三甲酯混合均匀,加热进行反应,得到粗产品,纯化,脱除溶剂,得到化合物2;
[0017]
将化合物2与正丁基锂、三丁基氯化锡混合均匀,进行反应,将反应产物纯化、脱除溶剂,得到化合物3;
[0018]
将化合物3与4-溴三苯胺、碳酸钾、四三苯基膦钯混合均匀,进行反应,得到化合物5;
[0019]
将化合物5与吡啶盐6在吡咯烷的催化下得到化合物tsvp;
[0020]
将化合物5与吡啶盐7在吡咯烷存在的条件下进行反应,得到产物不经纯化与六氟磷酸钾进行反应,得到tspy。
[0021]
本发明的合成路线如下描述:商业可得的硒酚,经过甲酰化的到2-甲酰硒酚1,2-甲酰硒酚1与原甲酸三甲酯反应上两个甲基保护得到缩醛2,缩醛2经过正丁基锂拔氢再与三丁基氯化锡发生反应后用盐酸水解脱除醛基保护得到锡试剂3,锡试剂3与4-溴三苯胺发生pd催化的偶联反应得到关键中间体醛5,醛5与吡啶盐6在吡咯烷催化下反应得到化合物tsvp。醛5在吡咯烷催化下与吡啶盐7发生缩合再用六氟磷酸钾处理得到tspy。具体反应路线图如图1所示
[0022][0023]
本发明的第三个方面,提供了上述的方法合成的聚集诱导发光材料在作为光动力治疗药物中的应用。
[0024]
本发明的第四个方面,提供了上述的聚集诱导发光材料在生物成像中的应用,其中,所述生物成像为细胞成像。
[0025]
本发明的有益效果
[0026]
(1)两个聚集诱导发光材料在光动力治疗中表现出了良好的治疗效果,可以杀灭癌细胞,同时能对a549细胞进行染色,文献中报道的类似化合物未见对a549细胞进行杀灭的实验报道。其中,tsvp还对金黄色葡萄球菌和耐药菌都具有较高的杀伤率。
[0027]
(2)本发明制备方法简单、实用性强,易于推广。
附图说明
[0028]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0029]
图1 tsvp和tspy的合成路线;
[0030]
图2化合物1的核磁共振氢谱;
[0031]
图3化合物1的核磁共振碳谱;
[0032]
图4化合物2的核磁共振氢谱;
[0033]
图5化合物2的核磁共振碳谱;
[0034]
图6化合物5的核磁共振氢谱;
[0035]
图7化合物5的核磁共振碳谱;
[0036]
图8化合物5的高分辨质谱;
[0037]
图9化合物tsvp的核磁共振氢谱;
[0038]
图10化合物tsvp的核磁共振碳谱;
[0039]
图11化合物tsvp的高分辨质谱;
[0040]
图12化合物tspy的核磁共振氢谱;
[0041]
图13化合物tspy的核磁共振碳谱;
[0042]
图14化合物tspy的高分辨质谱;
[0043]
图15 tsvp和tspy的紫外吸收图谱;
[0044]
图16 tsvp在不同比例四氢呋喃-水溶液中的荧光发射谱;
[0045]
图17 tspy在不同比例甲苯-二甲基亚砜溶液中的荧光发射谱
[0046]
图18 tsvp的细胞成像图;
[0047]
图19 tspy的细胞成像图;
[0048]
图20 tsvp对a549细胞的光动力治疗;
[0049]
图21 tspy对a549细胞的光动力治疗;
[0050]
图22 tsvp对金黄色葡萄球菌和耐药金黄色葡萄球菌的光动力治疗。
具体实施方式
[0051]
应该指出,以下详细说明都是例示性的,旨在对本发明提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0052]
本发明应用有机合成方法从商业可得的硒酚出发经过甲酰化、保护、锡化、金属催化的偶联、缩合等步骤得到含硒酚环的聚集诱导发光材料tsvp和tspy。
[0053][0054]
聚集诱导发光材料的合成方法,包括:
[0055]
将商业化的硒酚与三氯氧磷和n-甲基n-苯基甲酰胺混合均匀,室温搅拌反应,得到粗产品,后处理后脱除溶剂,纯化得到2-甲酰硒酚1;
[0056]
将2-甲酰硒酚1、甲醇、无水氯化铵、原甲酸三甲酯混合均匀,加热进行反应,得到粗产品,纯化,脱除溶剂,得到化合物2;
[0057]
将化合物2与正丁基锂、三丁基氯化锡混合均匀,进行反应,将反应产物纯化、脱除溶剂,得到化合物3;
[0058]
将化合物3与4-溴三苯胺、碳酸钾、四三苯基膦钯混合均匀,进行反应,得到化合物5;
[0059]
将化合物5与吡啶盐6在吡咯烷的催化下得到化合物tsvp;
[0060]
将化合物5与吡啶盐7在吡咯烷存在的条件下进行反应,得到产物不经纯化与六氟磷酸钾进行反应,得到tspy。
[0061]
在一些实施例中,所述2-甲酰硒酚、甲醇、无水氯化铵、原甲酸三甲酯的质量体积比为:1.89~2.0g:22.68~24ml:0.7~1.0g:2.34~2.5ml。
[0062]
在一些实施例中,所述化合物2、正丁基锂、三丁基氯化锡的摩尔比为1~2:1.2~
4:1.2~4。
[0063]
在一些实施例中,所述化合物3与4-溴三苯胺、碳酸钾、四三苯基膦钯的摩尔比为1~2:0.5~1:1.5~3:0.1~0.2。
[0064]
在一些实施例中,所述化合物5与吡咯烷、吡啶盐6的的摩尔比为1:0.1~2:1~1.2。
[0065]
在一些实施例中,所述化合物5与吡咯烷、吡啶盐7的的摩尔比为1:0.1~2:1~1.2。
[0066]
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
[0067]
实施例1
[0068][0069]
(1)二甲酰硒酚1的制备
[0070]
取2.71g(2.47ml,20.00mmol)n-甲基甲酰苯胺与3.07g(1.87ml,20.00mmol)三氯氧磷放入50ml支口烧瓶中,在氮保护下,磁力搅拌0.5h后,滴加硒酚2.50g(19.10mmol),在氮气氛下反应12h,tlc检测反应完全,将瓶内的混合物倒入500ml冰水化合物中。乙酸乙酯萃取水相3次,合并的有机相,用饱和的碳酸氢钠溶液洗涤1次。有机相用无水硫酸镁干燥,减压浓缩除去溶剂,粗产品经减压蒸馏得到淡黄色油状物2-甲酰硒酚(1)2.18g,产率为72%。1h nmr(500mhz,chloroform-d)δ9.83(d,j=1.1hz,1h),8.50(dt,j=5.4,1.2hz,1h),8.03(dd,j=3.9,1.2hz,1h),7.48(dd,j=5.4,3.9hz,1h);
13
c nmr(126mhz,chloroform-d)δ184.2,150.3,141.03,139.5,130.8。
[0071][0072]
(2)缩醛2的制备
[0073]
取2-甲酰硒酚7.02g(44mmol)加入200ml支口瓶中,加入100ml无水甲醇溶解,再加入无水氯化铵2.6g(49mmol)和原甲酸三甲酯8.69ml(7.82g,74mmol),在氮气保护下回流12h,tlc监测反应完全,减压浓缩除去甲醇,残余物用乙酸乙酯溶解,用1mol/l的氢氧化钠溶液洗涤,有机相再用饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,得到的无色粗产品经减压蒸馏后得到无色油状化合物缩醛2,5.23g,产率为58%。1h nmr(500mhz,chloroform-d)δ7.97(dd,j=5.0,1.8hz,1h),7.58
–
6.94(m,2h),5.59(s,1h),3.36(s,6h);
13
c nmr(126mhz,cdcl3)δ148.38,131.07,129.19,127.34,101.36,52.53.
[0074][0075]
(3)锡试剂3的制备
[0076]
缩醛2(615mg,3mmol)溶解于25ml支口瓶中,加入无水四氢呋喃12ml溶解,在-78℃下滴加正丁基锂溶液2.25ml(3.6mmol,1.6mol/l正己烷溶液)。滴加完毕后缓慢升至室温搅拌1h,然后在在-78℃下滴加三丁基氯化锡1.17g(3.6mmol,0.976ml),滴加完毕后搅拌搅拌过夜,向反应液中加入1mol/l的盐酸9ml,乙酸乙酯萃取有机相,分别用饱和碳酸氢钠、饱和食盐水洗涤,无水硫酸钠干燥,旋蒸除去溶剂,得到棕色油状物,未经进一步纯化直接用于下一步。
[0077][0078]
(4)醛5的制备
[0079]
上一步制备的锡试剂、4-溴三苯胺648mg(2mmol),碳酸钾552mg(4mmol)和四三苯基磷钯催化剂231mg(0.2mmol)加入25ml支口瓶中,加入甲苯8ml,n,n-二甲基甲酰胺2ml,于110℃搅拌过夜,反应完毕后旋蒸除去溶剂柱层析纯化得到黄色粉末652mg,产率81%.1h nmr(400mhz,chloroform-d)δ9.73(s,1h),7.94(d,j=4.2hz,1h),7.52
–
7.44(m,3h),7.36
–
7.27(m,4h),7.16
–
7.12(m,4h),7.09(td,j=7.2,1.2hz,2h),7.06
–
7.02(m,2h).
13
c nmr(101mhz,cdcl3)δ183.95,161.44,149.21,146.95,146.90,141.00,129.52,128.43,127.59,125.22,124.84,123.94,122.32.esi-hrms m/z:[m+h
+
]calculated for c
23h17
nose,calc 404.0548;measured 404.0546。
[0080][0081]
(5)tsvp的制备
[0082]
醛5(80.5mg,0.2mmol)与吡啶盐6(70.8mg,0.2mmol)放入25ml支口瓶,加入无水乙醇2ml,加入几滴吡咯烷,在氮气保护下室温搅拌12h,减压浓缩,柱层析纯化得到暗红色粉末97.5mg,产率66%。1h nmr(400mhz,dmso-d6)δ9.07(d,j=6.7hz,2h),8.35(d,j=15.7hz,1h),8.25(d,j=6.6hz,2h),7.68(d,j=4.3hz,1h),7.63(d,j=4.1hz,1h),7.61
–
7.56(m,2h),7.36(dd,j=8.4,7.3hz,4h),7.16
–
7.05(m,7h),6.96(d,j=8.8hz,2h),4.62(t,j=7.3hz,2h),3.52
–
3.44(m,2h),3.13(s,9h),2.47(dd,j=7.6,4.1hz,2h).
13
c nmr(101mhz,dmso)δ154.15,153.50,148.41,146.97,144.71,137.69,137.29,130.24,128.85,127.79,126.45,125.28,124.43,123.72,122.75,122.53,62.21,56.83,52.88,24.64.esi-hrms m/z:[m-2br-]calculated for c35h37n3se,calc 737.0519;measured 289.6087。
[0083]
[0084]
(6)tspy的制备
[0085]
醛5(80.5mg,0.2mmol)与吡啶盐7(47mg,0.2mmol)放入25ml支口瓶,加入无水乙醇2ml,加入几滴吡咯烷,在氮气保护下加热回流12h,减压浓缩,柱层析纯化得到暗红色粉末溶于5ml再加入饱和六氟磷酸钾溶液5ml,50加热搅拌3h后冷却到室温,过滤暗红色沉淀,沉淀用蒸馏水洗涤2次,用丙酮洗涤2次,真空干燥,得到棕红色粉末79mg,产率为62%。1h nmr(500mhz,dmso-d6)δ8.78(d,j=6.5hz,2h),8.19(d,j=15.7hz,1h),8.13(d,j=6.5hz,2h),7.66
–
7.52(m,4h),7.36(t,j=7.9hz,1h),7.19
–
7.07(m,6h),7.03(d,j=15.7hz,1h),6.96(d,j=8.7hz,2h),4.21(s,3h).
13
c nmr(126mhz,dmso)δ153.92,152.73,148.40,147.00,145.27,144.66,137.38,136.68,130.22,128.88,127.76,126.39,125.27,124.42,123.39,122.79,122.58,47.19.esi-hrms m/z:[m-f6p-]calculated for c
30 h
25 n
2 se,calc 493.1177;measured 493.1176。
[0086]
(7)紫外与荧光发射表征
[0087]
紫外测试:将tsvp用超纯水配置成浓度为10μm的溶液,tspy用二甲基亚砜配置为10μm的溶液,测试其紫外吸收曲线。
[0088]
荧光发射光谱测试:将tsvp用超纯水配置成浓度为10μm的溶液,tspy用二甲基亚砜配置为10μm的溶液,以480nm为激发波长测试荧光发射谱。
[0089]
(8)细胞成像实验
[0090]
在35mm培养皿中,盖玻片在37℃下培养细胞生长。a549细胞在一定温度下与特定染料一起集中繁殖一定时间。添加tsvp或tspy(500nm)后,将培养皿在室温下摇动几秒钟,然后盖玻片被取出。将tsvp或tspy标记细胞的玻片安装好后使用激光扫描共聚焦显微在488nm,5%激光功率(扫描速率为22.4s per框架)。发射滤光片为600-744nm。
[0091]
(9)a549细胞光动力治疗
[0092]
对于细胞毒性的测定一般采用mtt法进行。将细胞以每孔的密度6000-8000个接种于96孔板(costar,il,usa)上进行繁殖培养。在经过一夜培养过后将培养基每个孔中不同浓度的tsvp或tspy用100μl新鲜培养基代替。每个浓度分为光照和黑暗2组,光照组用日光灯照射10分钟,照射强度为10mw/cm2,黑暗组在黑暗中放置10分钟。然后将两组放于培养箱培养24h后,将mtt溶液向各个孔中分别加入10μl(在pbs中5mg/ml)。经过4h的繁殖后将100μl sds-hcl水溶液分别每个孔中。通过读板器记录595nm处繁殖4h每个孔的吸收(perkin elmer victor3tm)。每个试验用6个孔进行平行实验。
[0093]
化合物癌细胞杀伤率(对a549细胞)斯托克位移本技术的tspy95%192本技术的tsvp95%200
[0094]
(10)金黄色葡萄球菌及耐药菌的光动力治疗
[0095]
108个菌落(金黄色葡萄球菌或耐药金黄色葡萄球菌)形成单位分散在500μlpbs缓冲液中,并与不同浓度的tsvp共孵育10分钟,孵育结束后将菌液分散于pbs中并光照30分钟,对照组置于黑暗30分钟,用平板计数法定量细菌活力。细菌被稀释倍数为105。将100μl的菌液均匀涂抹在lb琼脂平板上,37℃培养18小时,定量拍照。对单菌落数量进行计数计算细菌的存活率。
[0096]
由此可知,本发明制备的聚集诱导发光材料tspy和tsvp不仅能对a549细胞进行染
色,对a549细胞也都具有较高的杀伤率,为该类癌症的治疗提供了新的方向,其中,tsvp还对金黄色葡萄球菌和耐药菌都具有较高的杀伤率,因此,有重要的研究价值。
[0097]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1