3,4-二氢-3-甲基-2H-1,4-苯并噁嗪及其制备方法与流程
3,4-二氢-3-甲基-2h-1,4-苯并噁嗪及其制备方法
技术领域
1.本发明属于精细化工制剂技术领域,涉及一种苯并噁嗪衍生物的制备,具体涉及一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪及其制备方法。
背景技术:
2.解草酮,又名解草嗪,化学名称为2,2-二氯-(3,4-二氢-3-甲基-2h-1,4-苯并噁嗪-4-基)乙酮。解草酮是二氯代乙酰胺类除草剂的解毒剂,在玉米中可以诱导谷胱甘肽巯基转移酶的形成,与异丙甲草胺轭合,从而降低异丙甲草胺对玉米的药害作用。解草酮常与其他除草剂配合使用,保护目标作物不受影响。
3.目前,解草酮通常由3,4-二氢-3-甲基-2h-1,4-苯并噁嗪与二氯乙酰氯在一定条件下制备得到。而3,4-二氢-3-甲基-2h-1,4-苯并噁嗪则通常通过两步制备获得(如吕程程,解草酯和解草酮的合成工艺研究[d],哈尔滨理工大学,2014;maria lopez-igesias,et al.chemoenzymatic asymmetric synthesis of 1,4-benzoxazine derivatives:application in the synthesis of a levofloxacin precursor[j].j.org.chem.2015,80,3815-3824):a、邻硝基苯酚与溴丙酮在催化剂作用下得到邻硝基苯氧丙酮;b、邻硝基苯氧丙酮在高温和pd/c催化剂的存在下与氢气发生还原反应,硝基被还原为胺基,同时与苯氧丙酮的羰基缩合脱去一分子水,分子内成环,环内双键继续被氢气还原而得到3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。制备3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的工艺需要使用具有强烈催泪作用、发泡作用及毒性大的溴丙酮,必须使用带压设备在高温高压下使原料与易燃爆的氢气发生一系列反应,安全措施要求高;另外,由于在密闭体系内反应,反应过程中无法通过取样检测对反应进行监控。
[0004]
此外,催化加氢常采用钯碳催化剂,此催化剂是金属钯微晶通过范德华力负载于活性炭表面的微孔内,其负载深度只有几十微米,在使用过程中任何摩擦撞击都可能会导致催化剂的磨损,变为更细小的催化剂颗粒分散在体系中,导致催化剂的活性降低和催化剂回收难度加大。活性炭表面微孔由于产物沉淀造成的堵塞也常常是催化活性降低的重要原因之一。因此在催化加氢过程中,昂贵的钯碳催化剂的消耗量大,造成生产成本居高不下。
[0005]
具体的,现有工艺主要存在如下问题:
[0006]
1、合成原料溴丙酮具有强烈催泪作用、发泡作用和毒性大,可能对操作人员的健康及环境产生影响;
[0007]
2、催化加氢过程需要使用带压设备,氢气在高温下易燃爆,安全措施要求高;由于反应在密闭体系中进行,反应过程中无法通过取样检测对反应进行监控;
[0008]
3、反应所需催化剂消耗量大,生产成本居高不下。
技术实现要素:
[0009]
为了克服上述现有技术的缺陷,本发明所要解决的技术问题是:提供一种3,4-二
氢-3-甲基-2h-1,4-苯并噁嗪的新制备方法,以及由制备方法所合成得到的3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。
[0010]
为了解决上述技术问题,本发明提供一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括在常压下进行如下步骤:
[0011]
s1、以邻乙酰氨基酚为原料,用卤代仲醇化合物键合酚羟基,获得2-(2-羟基-丙氧基)-乙酰苯胺,并水解获得2-(2-羟基-丙氧基)-苯胺;
[0012]
s2、2-(2-羟基-丙氧基)-苯胺在催化剂作用下胺基和仲羟基缩合成环,获得3,4-二氢-3-甲基-2h-1,4苯并噁嗪。
[0013]
进一步提供由前述制备方法所制备得到的3,4-二氢-3-甲基-2h-1,4苯并噁嗪。
[0014]
本发明的有益效果在于:本发明所提供的3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法所使用的设备均为常规设备,无需使用高压釜及氢气等危险源,制备过程中的原料及副产品对设备的腐蚀小,有利于保护操作人员的健康及生命安全,降低设备使用和维护成本。在特定催化剂的作用下,3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的产率高,纯度高,副产物少,副反应低,易于工业放大生产;所使用的树脂负载催化剂易回收,可反复使用多次而不降低催化效率;所使用的相转移催化剂成本低,绿色环保,对环境影响小。
附图说明
[0015]
图1所示为本发明在实施例2中2-(2-羟基-丙氧基)-乙酰苯胺的1h nmr谱图;
[0016]
图2所示为本发明在实施例2中2-(2-羟基-丙氧基)-苯胺的1h nmr谱图;
[0017]
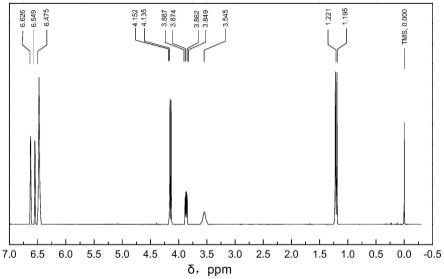
图3所示为本发明在实施例2中3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的1h nmr谱图。
具体实施方式
[0018]
为详细说明本发明的技术内容、所实现目的及效果,以下结合实施方式并配合附图予以说明。
[0019]
一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括在常压下进行如下步骤:
[0020]
s1、以邻乙酰氨基酚为原料,用卤代仲醇化合物键合酚羟基,获得2-(2-羟基-丙氧基)-乙酰苯胺,并水解获得2-(2-羟基-丙氧基)-苯胺;
[0021]
s2、2-(2-羟基-丙氧基)-苯胺在催化剂作用下胺基和仲羟基缩合成环,获得3,4-二氢-3-甲基-2h-1,4苯并噁嗪。
[0022]
其中,在本发明所提供的3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的整个反应过程中均允许在常压下进行反应,避免现有技术中高温高压下使用氢气所引发的燃爆风险。同时,反应使用的原料及副产物毒性低,对环境和操作人员健康影响小;反应在特定催化剂下进行,副反应少,得率高,制得的3,4-二氢-3-甲基-2h-1,4-苯并噁嗪纯度高。反应所使用的催化剂回收简单,可多次重复使用,催化效率下降不明显。
[0023]
其中,所述催化剂包括氧化锌和树脂负载催化剂。
[0024]
优选地,所述树脂负载催化剂由如下方法制备得到:
[0025]
以重量分数计,将100份大孔交联树脂mts9300烘干后,用甲苯煮沸,除
去线性聚合物及其他杂质;降温30~40℃后过滤收集大孔交联树脂,用无水乙醇置换甲苯;将置换后的大孔交联树脂重新加入反应釜中,以50%乙醇水分散后加热至40~60℃,将20~40g三氯化钌水合物溶于500g 50%乙醇水,在1~2h内,搅拌6~8h后降温,依次用50%乙醇水和无水乙醇洗涤至游离三氯化钌完全去除;以无水乙醇分散大孔交联树脂,加热至40~60℃,将240~480g三苯基膦溶于无水乙醇,在1~2h内缓慢滴入大孔交联树脂的无水乙醇分散液中,滴加完成后搅拌3~4h;降温至25~35℃,以无水乙醇洗涤至杂质完全去除后,室温真空干燥去除乙醇后,获得树脂负载催化剂。
[0026]
其中所述三氯化钌水合物中钌的质量含量为40%,三苯基膦的纯度为≥99%;
[0027]
具体的,在下文中,所述树脂负载催化剂由如下方法制备得到:将大孔交联树脂mts9300烘干后,取100g干树脂以1000g甲苯煮沸2小时,除去线性聚合物及其他杂质;降温至35℃过滤收集mts9300,在离子交换柱内以1000g无水乙醇置换出甲苯;将大孔交联树脂重新加入反应釜中,以500g 50%乙醇水分散,并加热至60℃;将25g三氯化钌水合物溶于500g 50%乙醇水,并缓慢滴入反应釜中,滴加完成后继续搅拌6小时,降温至25℃,依次以500g 50%乙醇水、500g无水乙醇洗涤至游离三氯化钌完全去除;以500g无水乙醇分散树脂,加热至40℃,将300g三苯基膦溶于1000g无水乙醇中,并缓慢滴入大孔交联树脂的无水乙醇分散液中,滴加完成后继续搅拌3小时;降温至25℃,以无水乙醇洗涤至杂质完全去除后,室温真空干燥除去树脂中的乙醇,得到树脂负载催化剂。
[0028]
在一种实施方式中,所述s1为:
[0029]
s11、以摩尔份数计,将1份邻乙酰氨基酚(cas号:614-80-2)加入反应釜,以15份去离子水分散,在1~2h内缓慢滴加入含1份碱的碱溶液,将1.5~3份卤代仲醇化合物溶于10份甲苯后加入反应釜中,再加入0.03~0.06份相转移催化剂,搅拌均匀后升温至70~80℃,保温4~12小时。降温至30~40℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯及过量的卤代仲醇减压蒸馏去除,得到2-(2-羟基-丙氧基)-乙酰苯胺;
[0030]
s12、以重量份数计,将1份2-(2-羟基-丙氧基)-乙酰苯胺溶于10份甲苯中,滴入5~8份碱溶液,加入0.03~0.06份相转移催化剂,搅拌并升温至80~85℃,保持6~8小时。降温至30~40℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯减压蒸馏去除,得到白色固体2-(2-羟基-丙氧基)-苯胺。
[0031]
在一种实施方式中,所述s2为:
[0032]
以重量份数计,将1份2-(2-羟基-丙氧基)-苯胺溶于6~8份甲苯,加入0.03~0.05份树脂负载催化剂和0.01~0.03份氧化锌,搅拌升温至100~110℃,蒸馏出反应的水分,蒸馏出的甲苯经分水器分离后返回反应釜中,保持8~10小时至反应无水生成后,降温至30~40℃,加入20~60ml 1mol/l盐酸,以700~800rpm的速度剧烈搅拌后静置,分离水层;有机层加入20~60ml 1mol/l氢氧化钠,以700~800rpm的速度剧烈搅拌后静置,分离水层;有机层经减压蒸馏除去甲苯,得到白色固体3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。
[0033]
在此步骤中,剧烈搅拌以促进盐酸与纳米氧化锌接触,从而溶解纳米氧化锌。其中纳米氧化锌在之后经油水分离,即将其从体系中除去。
[0034]
其中,所述碱为氢氧化钾或氢氧化钠;所述碱溶液的质量浓度为30~48%。
[0035]
在一种实施方式中,所述相转移催化剂为peg400或peg600。其中,peg400和peg600
在体系中状态较好,且水油界面粘度低。
[0036]
在一种实施方式中,所述卤代仲醇化合物为1-溴-2-丙醇或1-氯-2-丙醇。
[0037]
在一种实施方式中,所述氧化锌为纳米氧化锌,粒径为30~80nm,纯度为99%,在下文中,所述纳米氧化锌的典型牌号为xf106,购于江苏先丰纳米材料科技有限公司。
[0038]
3,4-二氢-3-甲基-2h-1,4-苯并噁嗪,由前述制备方法制备得到。
[0039]
本发明所述相转移催化剂不溶于使用的有机溶剂,可溶于水,在酸性或碱性条件下能够保持较高温度下稳定,在制备过程完成后可以经简单处理后除去,无毒无害,可降解,价格便宜,对环境影响小。
[0040]
本发明所述氧化锌的作用是使2-(2-羟基-丙氧基)-苯胺中的羟基易于脱氢,形成羰基化合物,同时氧化锌将脱离的氢转移至树脂负载催化剂表面。2-(2-羟基-丙氧基)-苯胺中的胺基电子云密度小,羟基脱氢形成的羰基化合物电子云密度大,两者相遇并反应,脱掉一分子水,发生环化反应,得到亚胺;之后吸附在催化剂表面的氢与亚胺加合,形成最终产物3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。氧化锌和树脂负载催化剂恢复原状,继续催化反应,因而氧化锌与树脂负载催化剂产生协同作用,降低了环化的活化能,提高了对所述反应的催化能力,所得产物产率高,纯度高。
[0041]
本发明所述树脂负载催化剂为固体催化剂,其催化效率高,在反应完成后过滤或离心即可排出体系,避免了萃取、蒸馏等复杂的处理过程。同时,回收的树脂负载催化剂效率仍然较高,在使用十次后催化效率无明显降低,因此,可降低生产成本,提高效率。所述氧化锌在反应完成后可加入稀盐酸溶解于水中,通过油水分离即可排出体系,不影响目标物纯度。
[0042]
实施例1
[0043]
一种树脂负载催化剂由如下方法制备得到:
[0044]
将大孔交联树脂mts9300烘干后,取100g干树脂以1000g甲苯煮沸2小时,除去线性聚合物及其他杂质;降温至35℃过滤收集mts9300,在离子交换柱内以1000g无水乙醇置换出甲苯;将大孔交联树脂重新加入反应釜中,以500g 50%乙醇水分散,并加热至60℃;将25g三氯化钌水合物(钌的质量含量为40%)溶于500g 50%乙醇水,并缓慢滴入反应釜中,滴加完成后继续搅拌6小时,降温至25℃,依次以500g 50%乙醇水、500g无水乙醇洗涤至游离三氯化钌完全去除;以500g无水乙醇分散树脂,加热至40℃,将300g三苯基膦溶于1000g无水乙醇中,并缓慢滴入树脂的无水乙醇分散液中,滴加完成后继续搅拌3小时;降温至25℃,以无水乙醇洗涤至杂质完全去除后,室温真空干燥除去树脂中的乙醇,得到树脂负载催化剂。
[0045]
实施例2
[0046]
一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括如下步骤:
[0047]
s1、将151.2g邻乙酰氨基酚加入反应釜,以270g去离子水分散,在2h内缓慢滴加入186.7g 30% koh溶液,将208.5g 1-溴-2-丙醇溶于920g甲苯后加入反应釜中,再加入18g相转移催化剂peg600,搅拌均匀后升温至80℃,保温4小时。降温至30℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯及过量的1-溴-2-丙醇减压蒸馏去除,得到2-(2-羟基-丙氧基)-乙酰苯胺。2-(2-羟基-丙氧基)-乙酰苯胺的收率为93.31%,纯度为95.55%。其红外谱图(1h nmr谱图)如图1所示。各氢化学位
移δ:1.207-1.233(双峰,3h,ch3),2.024(单峰,3h,coch3),2.170(单峰,h,oh),3.903-3.939(四重峰,1h,ch),4.100-4.119(双峰,2h,ch2),6.694(单峰,h,ar-h),6.791(单峰,1h,ar-h),6.941(单峰,h,ar-h),7.540(单峰,h,ar-h),7.853(单峰,h,conh)。
[0048]
s2、将100g 2-(2-羟基-丙氧基)-乙酰苯胺溶于1000g甲苯中,滴入600g 40%naoh溶液,加入18g相转移催化剂peg600,搅拌并升温至85℃,保持6小时。降温至40℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯减压蒸馏去除,得到白色固体2-(2-羟基-丙氧基)-苯胺。2-(2-羟基-丙氧基)-苯胺的收率为97.17%,纯度为96.35%。其红外谱图(1h nmr谱图)如图2所示。各氢化学位移δ:1.207-1.234(双峰,3h,ch3),2.187(单峰,1h,oh),3.711(单峰,2h,nh2),3.887-3.924(四重峰,1h,ch),4.085-4.103(双峰,2h,ch2),6.630(单峰,h,ar-h),6.532(单峰,2h,ar-h),6.605(单峰,h,ar-h)。
[0049]
s3、将80g 2-(2-羟基-丙氧基)-苯胺溶于480g甲苯,加入4g树脂负载催化剂和2.4g纳米氧化锌xf106,搅拌升温至105℃,蒸馏出反应的水分,蒸馏出的甲苯经分水器分离后返回反应釜中,保持9小时至反应无水生成后,降温至30℃,加入60ml 1mol/l盐酸,以700rpm的速度剧烈搅拌后静置,分离水层;有机层加入60ml 1mol/l氢氧化钠,以700rpm的速度剧烈搅拌后静置,分离水层;有机层经蒸馏除去甲苯,得到白色固体3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的总收率为84.77%,纯度为97.18%。其红外谱图(1h nmr谱图)如图3所示。各氢化学位移δ:1.195-1.221(双峰,3h,ch3),3.545(单峰,1h,nh),3.849-3.887(四重峰,1h,ch),4.135-4.152(双峰,2h,ch2),6.475(单峰,2h,ar-h),6.549(单峰,1h,ar-h),6.626(单峰,h,ar-h)。
[0050]
实施例3
[0051]
一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括如下步骤:
[0052]
s1、将151.2g邻乙酰氨基酚加入反应釜,以270g去离子水分散,在1h内缓慢滴加入83.4g 48% naoh溶液,将283.5g 1-氯-2-丙醇溶于920g甲苯后加入反应釜中,再加入24g相转移催化剂peg400,搅拌均匀后升温至70℃,保温12小时。降温至40℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯及过量的1-氯-2-丙醇减压蒸馏去除,得到2-(2-羟基-丙氧基)-乙酰苯胺。其收率为94.14%,纯度为95.17%。
[0053]
s2、将100g 2-(2-羟基-丙氧基)-乙酰苯胺溶于1000g甲苯中,滴入800g 30%koh溶液,加入24g相转移催化剂peg400,搅拌并升温至80℃,保持8小时。降温至35℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯减压蒸馏去除,得到白色固体2-(2-羟基-丙氧基)-苯胺。其收率为95.89%,纯度为96.78%。
[0054]
s3、将50g 2-(2-羟基-丙氧基)-苯胺溶于350g甲苯,加入2g树脂负载催化剂和0.5g纳米氧化锌xf106,搅拌升温至110℃,蒸馏出反应的水分,蒸馏出的甲苯经分水器分离后返回反应釜中,保持8小时至反应无水生成后,降温至30℃,加入20ml 1mol/l盐酸,以800rpm的速度剧烈搅拌后静置,分离水层;有机层加入20ml 1mol/l氢氧化钠,以800rpm的速度剧烈搅拌后静置,分离水层;有机层经蒸馏除去甲苯,得到白色固体3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。其总收率为84.06%,纯度为97.14%。
[0055]
实施例4
[0056]
一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括如下步骤:
[0057]
s1、将151.2g邻乙酰氨基酚加入反应釜,以270g去离子水分散,在1.5h内缓慢滴加140g 40%koh,将280g 1-溴-2-丙醇溶于920g甲苯后加入反应釜中,再加入21g相转移催化剂peg600,搅拌均匀后升温至75℃,保温8小时。降温至35℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯及过量的1-溴-2-丙醇减压蒸馏去除,得到2-(2-羟基-丙氧基)-乙酰苯胺。其收率为93.38%,纯度为96.19%。
[0058]
s2、将100g 2-(2-羟基-丙氧基)-乙酰苯胺溶于1000g甲苯中,滴入500g 48%naoh溶液,加入21g相转移催化剂peg600,搅拌并升温至83℃,保持7小时。降温至40℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯减压蒸馏去除,得到白色固体2-(2-羟基-丙氧基)-苯胺。其收率为98.46%,纯度为96.31%。
[0059]
s3、将50g 2-(2-羟基-丙氧基)-苯胺溶于400g甲苯,加入1.5g树脂负载催化剂和1g纳米氧化锌xf106,搅拌升温至100℃,蒸馏出反应的水分,蒸馏出的甲苯经分水器分离后返回反应釜中,保持10小时至反应无水生成后,降温至35℃,加入30ml 1mol/l盐酸,以750rpm的速度剧烈搅拌后静置,分离水层;有机层加入30ml 1mol/l氢氧化钠,以750rpm的速度剧烈搅拌后静置,分离水层;有机层经蒸馏除去甲苯,得到白色固体3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。其总收率为85.98%,纯度为96.95%。
[0060]
实施例5
[0061]
一种3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的制备方法,包括如下步骤:
[0062]
s1、将151.2g邻乙酰氨基酚加入反应釜,以270g去离子水分散,在2h内缓慢滴加入140g 40% koh,将236g 1-氯-2-丙醇溶于920g甲苯后加入反应釜中,再加入16g相转移催化剂peg400,搅拌均匀后升温至78℃,保温6小时。降温至30℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯及过量的1-氯-2-丙醇压蒸馏去除,得到2-(2-羟基-丙氧基)-乙酰苯胺。其收率为92.85%,纯度为96.08%。
[0063]
s2、将100g 2-(2-羟基-丙氧基)-乙酰苯胺溶于1000g甲苯中,滴入720g 36%koh溶液,加入16g相转移催化剂peg400,搅拌并升温至85℃,保持6小时。降温至35℃,静置,弃去下层水层,以去离子水洗涤上层有机层,弃去下层水层,将上层有机相中的有机溶剂甲苯减压蒸馏去除,得到白色固体2-(2-羟基-丙氧基)-苯胺。其收率为96.91%,纯度为95.94%。
[0064]
s3、将50g 2-(2-羟基-丙氧基)-苯胺溶于320g甲苯,加入1.2g树脂负载催化剂和1.5g纳米氧化锌xf106,搅拌升温至105℃,蒸馏出反应的水分,蒸馏出的甲苯经分水器分离后返回反应釜中,保持9小时至反应无水生成后,降温至40℃,加入40ml 1mol/l盐酸,以800rpm的速度剧烈搅拌后静置,分离水层;有机层加入40ml 1mol/l氢氧化钠,以800rpm的速度剧烈搅拌后静置,分离水层;有机层经蒸馏除去甲苯,得到白色固体3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。其总收率为93.28%,纯度为84.02%。
[0065]
实施例6
[0066]
采用与实施例4相同的技术方案,将其所使用的树脂负载催化剂进行回收后继续催化反应。其中,树脂负载催化剂重复10次,反应得到3,4-二氢-3-甲基-2h-1,4-苯并噁嗪。
10次重复所得到3,4-二氢-3-甲基-2h-1,4-苯并噁嗪的产物收率和纯度如表1所示。
[0067]
表1
[0068]
重复次数收率(%)纯度(%)185.9897.21285.6597.05385.7396.89485.5896.64585.2296.95685.4997.32785.7496.77885.2596.31984.9796.831084.7196.90
[0069]
从表1可以看出,本发明所提供的树脂负载催化剂在使用十次后其催化效率无明显下降,即有效降低生产成本,提高生产效率。
[0070]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1