一种遗传改造的里氏木霉工程菌株及其制备方法和应用与流程
1.本发明属于生物工程技术领域,具体涉及微生物的遗传物质的改造,具体为真菌里氏木霉的基因工程改造。
背景技术:
2.随着石化原料短缺和环境污染日趋严峻,对绿色环保新能源、新材料的需求也日益迫切。生物质作为唯一可再生、可循环利用的原料,在取代石油生产新材料的产业开发中具有不可取代的地位。植物木质纤维素材料,如废弃农作物秸秆,是最为丰富的生物质原料来源,仅中国就年产数亿吨之多。经过预处理及纤维素酶降解,纤维素和半纤维素转化出的单糖可以发酵成为小分子产物如乳酸,而这些小分子可以进而合成材料产品,如聚乳酸,即可降解塑料。这样的新材料生物炼制产业能够满足迫切的市场需求,并解决石化炼制带来环境问题,如“白色污染”。
3.在木质纤维素的利用过程中,纤维素酶的降解糖化是关键的一步,但目前纤维素酶的生产成本高、价格贵,不能满足生物炼制产业发展的生产和盈利需求,成为生物质新材料实现产业化的重要瓶颈。里氏木霉rut-c30是现今应用最为广泛的纤维素酶生产菌。目前,对于里氏木霉生产纤维素酶工艺的研发已经较为成熟,要进一步提高纤维素酶的酶活及产量,需从优化源头菌株的角度出发,利用合成生物学的原料以及基因编辑等先进技术手段,设计并遗传改造出产纤维素酶能力更强的里氏木霉工程菌株。
4.里氏木霉诱导产生的纤维素酶组分中,包含外切葡聚糖酶、内切葡聚糖酶以及β葡萄糖苷酶。纤维素酶降解纤维素过程中,外切酶及内切酶协同将纤维素降解为纤维寡糖(主要为纤维二糖),之后β-葡萄糖苷酶将纤维二糖降解为终产物葡萄糖,用于下游新能源、新材料的生产。三种酶组分分别受不同基因的控制,其中内外切酶基因编码的酶占到整个纤维素酶系的95%,而β葡萄糖苷酶基因编码的酶只有不到5%。β葡萄糖苷酶的缺乏严重制约了纤维素的降解,目前多通过额外补充β酶的方式来弥补酶系中的不足,但这样不仅增加了酶解成本,也增加了人工操作方面的复杂性。因此,提高β葡萄糖苷酶的表达水平,优化里氏木霉生产纤维素酶的酶系组成是必要的。
技术实现要素:
5.为克服现有技术存在的上述的缺陷,本发明将来源于黑曲霉的bgl1b克隆出来,并构建出该基因的过表达载体,于里氏木霉里过表达,弥补了里氏木霉所产β葡萄糖苷酶不充分,纤维素复合酶系不够完善的缺陷。另外,本发明针对里氏木霉,构建了纤维素酶相关基因的转录抑制因子rce1的crispr-cas9的敲除载体,通过该载体将rce1基因成功敲除,结果进一步提高了纤维素酶相关基因的表达水平,验证了rce1基因对于纤维素酶基因表达水平的抑制作用,确认其作为转录负调控因子的可能性。最后,本发明将经过双重改造的里氏木霉工程菌株筛选出来,从产纤维素酶的工艺角度出发对其进行针对性的工艺优化,进一步提高了工程菌株生产纤维素酶的能力,从微观及宏观角度出发,为工业生产纤维素酶提供
了优良的工程菌株。提供如下的技术方案:
6.本发明的第一个方面是提供一种基因改造的里氏木霉的方法,所述的方法是敲除内源性基因rce1的表达,同时过表达外源bgl1b。优选地,所述的里氏木霉为rut-c30;优选地,所述的外源bgl1b来源于黑曲霉,优选的,所述的bgl1b的编码序列为seq id no:1所示。优选地,所述的敲除内源性基因rce1的表达的方法包括但不限于同源重组、rna干扰、zfn、talens、crispr-cas9,优选的,所述的基因编辑采用的是crispr-cas9系统,其中sgrna的序列为seq id no:2所示;更优选的,所述的crispr-cas9系统是将cas9表达框与sgrna克隆进同一个表达质粒中转化宿主细胞;更优选的,所述的质粒还包括了启动子、抗性基因片段、终止子元件;其中,sgrna序列相关元件以如下方式连接获得:hh ribozyme-grna-rce1(rut-c30)-sgrna_backbone-hdv ribozyme片段通过碱基合成方法获得,其中hh ribozyme序列如seq id no:3所示;sgrna_backbone序列如seq idno:4所示;hdv ribozyme序列如seq id no:5所示。
7.在另外的优选方案中,所述的方法中还包括检测基因改造的里氏木霉相关基因,其中所述的相关基因包括内源性的rce1和外源的bgl1b,将rce1基因敲除成功和bgl1b转化成功的作为候选的菌株进行酶活检测。
8.在另外的优选方案中,所述的方法还包括使用rt-qpcr方法测定相关基因:外切酶基因cbh1、内切酶基因egl1、β葡萄糖苷酶基因bgl1的表达量,最终获得滤纸酶活最高且酶基因表达量最高的工程菌株。
9.本发明的第二个方面提供上述第一方面所述的方法产生的基因工程菌。
10.本发明的第三个方面提供第二个方面所述的基因工程菌在发酵生产纤维素酶中的应用。在一个具体的实施例中,所述的发酵生产纤维素酶的条件包括显示在种子培养25~35h,移种量为10~15%,初始ph为5~6。在一个优选的实施例中,所述的发酵为工业化生产发酵,发酵罐容积为200l~500l。
11.本发明的有益效果在于:
12.1)以常规里氏木霉为出发菌株,经过基因工程改造敲除内源性的rce1,过表达来源于黑曲霉的bgl1b后获得的基因工程菌tr-hh01,对其发酵获得的酶液来酶解秸秆底物时,会获得更高的葡萄糖组分、木糖组分及酶解总糖,同时纤维二糖组分有明显降低,说明经过遗传改造,工程菌株tr-hh01可被诱导产生更高的纤维素酶、木聚糖酶、及纤维素酶中的β-葡萄糖苷酶组分,从而不再需要额外添加外源的β-葡萄糖苷酶,大大降低了工业应用纤维素酶的成本。
13.2)工程菌株tr-hh01,以在获得的最佳发酵条件进行300l发酵罐的放大验证,本发明开发的工程菌株及发酵工艺具有明显优势,可为纤维素酶的工业生产提供良好的支撑。
附图说明
14.图1质粒pbargpe1-bgl1b的结构示意图;
15.图2质粒pcrispr-rce1的结构示意图;
16.图3pbargpe1-bgl1b转化里氏木霉rut-c30菌株pcr验证结果;
17.图4pcrispr-rce1转化里氏木霉rut-30菌株pcr验证结果;
18.图5pcr验证rce1基因是否敲除成功;
19.图6不同发酵条件下的滤纸酶活柱状图;
20.图7工业化发酵tr-hh01菌株的产酶情况。
具体实施方式
21.以下通过参考示范性实施例,本发明的目的和功能以及用于实现这些目的和功能的方法将得以阐明。然而,本发明并不受限于以下所公开的示范性实施例;可以通过不同形式来对其加以实现。说明书的实质仅仅是帮助相关领域技术人员综合理解本发明的具体细节。
22.实施例1过表达载体pbargpe1-bgl1b构建
23.ppdc启动子序列(1534bp)及tpdc终止子序列(1030bp)来源里氏木霉,bgl1b基因(gene id:4980363)来源于黑曲霉,序列如seq id no:1所示,将上述片段通过pcr技术克隆出来,构建到骨架载体pbargpe1(购自addgene)中,得到bgl1b的过表达质粒pbargpe1-bgl1b,质粒全序列8952bp,所述质粒的结构示意图如图1所示。
24.实施例2crispr-cas9基因敲除载体pcrispr-rce1构建
25.1、rce1基因的sgrna序列设计
26.从ncbi上获得rce1基因序列,并标记出cds区(sequence coding for aminoacids in protein,是编码一段蛋白产物的序列)。利用http://grna.ctegd.uga.edu/网站对rce1基因的第一个cds区进行sgrna的设计,本发明设计的sgrna序列为:tgggtgagtgcacctgatgatgg(seq id no:2),标记为grna-rce1(rut-c30)。
27.2、rce1的crispr-cas9基因敲除载体构建
28.抗性筛选片段获得:gpda启动子片段(gpda promoter,2129bp)、潮霉素抗性基因片段(hph,1144bp)、trpc终止子片段(trpc terminator,764bp)来源于pan7-1质粒(购自addgene),将上述gpda promoter-hph-trpc terminator片段通过pcr及gibson assembly技术克隆组装待用。
29.cas9表达框片段获得:ptef启动子片段(ptef promoter,886bp)、cas9片段(4104bp)、ptef终止子片段(ptef terminator,489bp)来源于质粒pfc334(购自addgene),上述片段通过pcr及gibson assembly技术克隆组装得到。
30.sgrna相关序列元件组装:hh ribozyme-grna-rce1(rut-c30)-sgrna_backbone-hdv ribozyme片段通过碱基合成方法获得,其中hh ribozyme序列如seq id no:3所示;sgrna_backbone序列如seq idno:4所示;hdv ribozyme序列如seq id no:5所示。此外,质粒上固有片段(例如ampr、ori等片段)也通过常规分子克隆技术获得。最后将上述几个片段通过酶连方式连接到一起,得到rce1基因的crispr-cas9基因敲除载体pcrispr-rce1,质粒全序列11762bp(图2)。
31.以上质粒使用天根生化科技有限公司的质粒小提试剂盒(离心柱型,dpp103-02)提取,用于后续转化。
32.实施例3质粒转化宿主及筛选
33.1载体pbargpe1-bgl1b转化里氏木霉rut-c30菌株
34.1)转化:将步骤2.1中构建的载体pbargpe1-bgl1b转化里氏木霉rut-c30菌株(常规步骤),涂布于抗性筛选培养基,抗性筛选标记为博来霉素(blpr,工作浓度100ug/ml)。
35.其中,以上抗性筛选培养基配方为:1m d-山梨糖醇,20g/l葡萄糖,15g/l kh2po4,5g/l(nh4)2so4,0.6g/l mgso4·
7h2o,0.6g/l cacl2,0.005g/lfeso4·
7h2o,0.0016g/l mnso4·
h2o,0.0014g/l znso4·
7h2o,0.002g/l cocl2,20g/l琼脂粉。
36.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入博来霉素(blpr,工作浓度100ug/ml)和氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
37.2)扩繁:将抗性筛选培养基上长出的单克隆转化株用无菌牙签挑取转移至不含潮霉素的木霉琼脂培养基扩繁,生长7天后,加10ml无菌水刮取得到孢子悬浮液储存于4℃冰箱待用。
38.以上木霉琼脂培养基配方为:10g/l葡萄糖,2g/l玉米浆,2.8g/lkh2po4,3.92g/l(nh4)2so4,0.84g/l mgso4·
7h2o,0.84g/l尿素,2g/l吐温-80,0.014g/l feso4·
7h2o,0.00436g/l mnso4·
h2o,0.00392g/lznso4·
7h2o,0.0056g/l cocl2,20g/l琼脂;配制好之后,用2m的naoh溶液调节ph至4.8。
39.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
40.3)木霉菌种培养:将转化株和里氏木霉rut-c30菌株的孢子悬浮液接种于木霉菌种培养基(100ml),孢子接种量为10^9cell/l,接种结束过夜培养,得到转化株的菌丝。
41.以上木霉菌种培养基配方为:12g/l葡萄糖,0.7g/l玉米浆,1.96g/lkh2po4,1.372g/l(nh4)2so4,0.294g/l mgso4·
7h2o,0.294g/l尿素,0.0049g/l feso4·
7h2o,0.0015g/l mnso4·
h2o,0.0014g/l znso4·
7h2o,0.002g/lcocl2。
42.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
43.dna提取:使用天根生化科技有限公司的快捷型植物基因组dna提取系统(dp-321)提取过夜培养的木霉菌丝的dna,操作参照试剂盒说明书。
44.引物设计:验证质粒转化成功与否的相关引物设计为(5
’‑3’
):
45.bgl1b-f:tgcctccggactttctctgg(seq id no:6);
46.bgl1b-r:gctatctccctctgcgctctt(seq id no:7)。
47.琼脂糖凝胶电泳:以质粒(以p代替)为阳性对照,里氏木霉rut-c30(以w代替)为阴性对照,用bgl1b基因上设计的引物bgl1b-f和bgl1b-r对转化株dna和对照dna进行pcr扩增和琼脂糖凝胶电泳(常规步骤),本发明一共挑取9个转化株,其中4个存在目的条带(图3),证明4株转化成功。
48.对以上4株阳性转化株(分别命名为a1、a2、a3、a4)进行摇瓶发酵验证,发酵结束测定纤维素酶活,本发明以滤纸酶活(filter paper activity,fpa)定量纤维素酶活,并采用国际理论与应用化学联合会(international union of pure and applied chemistry,iupac)推荐的酶活标准测定方法进行测定,第七天酶活结果分别是4.81、6.21、5.1、3.7fpu/ml,将测定酶活最高的转化株a2,进行扩繁,待用。
49.2载体pcrispr-rce1转化转化里氏木霉rut-c30菌株
50.转化:将步骤2.2中构建的载体pcrispr-rce1转化3.1中获得的a2菌株(常规步骤),涂布于抗性筛选培养基,抗性筛选标记为潮霉素(hph,工作浓度100ug/ml)。
51.其中,以上抗性筛选培养基配方为:1m d-山梨糖醇,20g/l葡萄糖,15g/l kh2po4,5g/l(nh4)2so4,0.6g/l mgso4·
7h2o,0.6g/l cacl2,0.005g/lfeso4·
7h2o,0.0016g/l mnso4·
h2o,0.0014g/l znso4·
7h2o,0.002g/l cocl2,20g/l琼脂粉。
52.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入潮霉素(hph,工作浓度150ug/ml)和氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
53.扩繁:将抗性筛选培养基上长出的单克隆转化株用无菌牙签挑取转移至不含潮霉素的木霉琼脂培养基扩繁,生长7天后,加10ml无菌水刮取得到孢子悬浮液储存于4℃冰箱待用。
54.以上木霉琼脂培养基配方为:10g/l葡萄糖,2g/l玉米浆,2.8g/lkh2po4,3.92g/l(nh4)2so4,0.84g/l mgso4·
7h2o,0.84g/l尿素,2g/l吐温-80,0.014g/l feso4·
7h2o,0.00436g/l mnso4·
h2o,0.00392g/lznso4·
7h2o,0.0056g/l cocl2,20g/l琼脂;配制好之后,用2m的naoh溶液调节ph至4.8。
55.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
56.木霉菌种培养:将转化株和里氏木霉rut-c30菌株的孢子悬浮液接种于木霉菌种培养基(100ml),孢子接种量为10^9cell/l,接种结束过夜培养,得到转化株的菌丝。
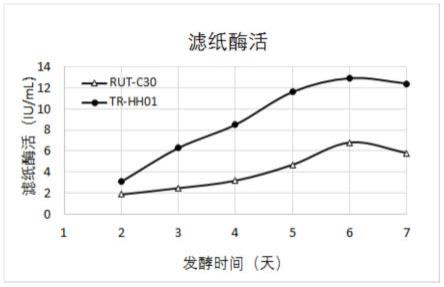
57.以上木霉菌种培养基配方为:12g/l葡萄糖,0.7g/l玉米浆,1.96g/lkh2po4,1.372g/l(nh4)2so4,0.294g/l mgso4·
7h2o,0.294g/l尿素,0.0049g/l feso4·
7h2o,0.0015g/l mnso4·
h2o,0.0014g/l znso4·
7h2o,0.002g/lcocl2。
58.高压蒸汽灭菌锅中灭菌(121℃,25min);待温度冷却至45-50℃(以不烫手为宜),于超净工作台中加入氨苄青霉素(终浓度100μg/ml),后倒塑料培养皿冷却待用。
59.dna提取:使用天根生化科技有限公司的快捷型植物基因组dna提取系统(dp-321)提取过夜培养的木霉菌丝的dna,操作参照试剂盒说明书。
60.引物设计:验证质粒转化成功与否的相关引物设计为(5
’‑3’
):
61.crispr-f:tcagagaggtcaaggtcatc(seq id no:8);
62.crispr-r:ctctccatgatggtgattcc(seq id no:9)。
63.琼脂糖凝胶电泳:以质粒(以p代替)为阳性对照,里氏木霉rut-c30(以w代替)为阴性对照,用pcrispr-rce1质粒上设计的引物crispr-f和crispr-r对转化株dna和对照dna进行pcr扩增和琼脂糖凝胶电泳(常规步骤),本发明一共挑取12个转化株,其中7个存在目的条带(图4),证明此7株转化成功。
64.以敲除基因rce1上跨sgrna的区域为目标片段,引物对得到的阳性转化子dna进行扩增,并以原始菌株rut c30作为阳性对照,对扩增产物进行琼脂糖凝胶电泳(图5)。图中泳道m为marker,泳道1、2、3、4、5、6、7分别为7个转化株基因组扩增条带,泳道w为原始菌株rut c30扩增条带,其中有5株转化株无目的条带,其余转化菌株与原始菌株有条带,故此5株转化株的rce1基因敲除成功。
65.对以上5株转化株进行摇瓶发酵,在发酵第三天测定滤纸酶活,并使用rt-qpcr方法测定相关基因:外切酶基因cbh1、内切酶基因egl1、β葡萄糖苷酶基因bgl1的表达量,最终获得滤纸酶活最高且酶基因表达量最高的工程菌株,将其命名为tr-hh01。
66.实施例4tr-hh01菌株发酵及酶活测定
67.1)tr-hh01摇瓶发酵过程如下:摇瓶种子和摇瓶发酵培养基体系均为100ml,摇瓶种子的接孢量为10^7cell/l,种子培养时间为18h,种子至发酵的移种量为10%,种子及发酵过程的培养条件为30℃、180rpm,ph不控制,发酵培养时间为3天。
68.以上摇瓶种子培养基组分为:葡萄糖22g/l,玉米浆1.7g/l,磷酸二氢钾2g/l,硫酸铵0.5g/l,硫酸镁0.83g/l、七水硫酸亚铁0.005g/l,一水硫酸锰0.0015g/l,七水硫酸锌0.0014g/l,氯化钴0.002g/l,加水定容至100毫升。配制好之后于121℃,25min条件下灭菌消毒,冷却待用;
69.以上摇瓶发酵培养基组分为:汽爆玉米秸秆2%,玉米浆粉2g/l,磷酸二氢钾1g/l,硫酸铵2g/l,七水硫酸镁3g/l,七水硫酸亚铁0.005g/l,一水硫酸锰0.0015g/l,七水硫酸锌0.0014g/l,氯化钴0.002g/l,加水定容至100ml,配制好之后于121℃,25min条件下灭菌消毒,冷却待用;
70.2)检测相关酶表达量
71.rna的提取、反转录、及rt-qpcr分布按照easyspin plus植物rna快速提取试剂盒、takara的反转录试剂盒rr047a、rt-qpcr的试剂盒rr420a操作。内参基因选用sar1。
72.以上rt-qpcr过程中使用到的相关引物设计如表1:
73.表1所用引物序列
74.引物名称引物序列(5
’‑3’
)qsar1-ftggatcgtcaactggttctacga(seq id no:10)qsar1-rgcatgtgtagcaacgtggtcttt(seq id no:11)qcbh1-facgagttctctttcgatgttgatg(seq id no:12)qcbh1-rcggtgttggtgggatacttg(seq id no:13)qeg1-factacaactcgtgcaccgtc(seq id no:14)qeg1-rccgaggtcgtgacgcccgag(seq id no:15)qbgl1b-fatagatacggtcagcctaag(seq id no:16)qbgl1b-rccatataagccctcacattc(seq id no:17)
75.tr-hh01摇瓶发酵3天酶活fpa=4.01iu/ml,rut-c30菌株摇瓶发酵3天酶活fpa=2.01iu/ml;tr-hh01相对于rut-c30菌株的cbh1、egl1、bgl1基因的相对表达量分别为:59、43、78。
76.实施例5产纤维素酶工程菌株的发酵工艺优化
77.将经过合成方法获得的工程菌株tr-hh01进行中试发酵工艺探索。
78.种子培养时间及移种量:将孢子从平板培养基中收集下来(浓度10^8cell/ml),接种到种子培养基内,接种量为10^9cell/l,在180rpm、30℃的条件下,分别培养18h、24h、30h、36h,之后将种子液移种到发酵培养基,移种量分别为8%、10%、12%、15%。
79.其中,种子罐培养基组分为:葡萄糖22g/l,玉米浆1.7g/l,磷酸二氢钾2g/l,硫酸铵0.5g/l,硫酸镁0.83g/l、七水硫酸亚铁0.005g/l,一水硫酸锰0.0015g/l,七水硫酸锌0.0014g/l,氯化钴0.002g/l,加水定容至1l。配制好之后于121℃,25min条件下灭菌消毒,冷却待用;
80.发酵培养基组分为:汽爆玉米秸秆2%,玉米浆粉2g/l,磷酸二氢钾1g/l,硫酸铵
2g/l,七水硫酸镁3g/l,七水硫酸亚铁0.005g/l,一水硫酸锰0.0015g/l,七水硫酸锌0.0014g/l,氯化钴0.002g/l,加水定容至10l,配制好之后于121℃,25min条件下灭菌消毒,冷却待用;
81.发酵过程ph控制:发酵过程中,温度设置为30℃,do设置为30%,初始ph分别设置为4.5、5.5。并从第2天开始测定滤纸酶活,直至第7天产酶下罐。
82.本发明将每个条件下所测得的最高酶活加以对比(图6),显示在种子培养30h,移种量为12%,初始ph为5.5时,可使工程菌株tr-hh01发酵获得最高的滤纸酶活10.5iu/ml。
83.实施例6产纤维素酶工程菌株的放大培养
84.上述实施例方法获得工程菌株tr-hh01,以实施例5获得的最佳发酵条件进行300l发酵罐的放大验证,并以出发菌株里氏木霉rut-c30进行酶活对照,从第2天开始取样测定滤纸酶活,直至发酵第7天下罐。结果发现(图7),本发明开发的工程菌株及发酵工艺具有明显优势,可为纤维素酶的工业生产提供良好的支撑。
85.取两菌株发酵结束后的酶液,酶解蒸汽爆破预处理后的秸秆,对酶解糖进行对比,结果发现(表2),相对于出发菌株rut-c30来说,以工程菌株tr-hh01发酵获得的酶液来酶解秸秆底物时,会获得更高的葡萄糖组分、木糖组分及酶解总糖,同时纤维二糖组分有明显降低,说明经过遗传改造,工程菌株tr-hh01可被诱导产生更高的纤维素酶、木聚糖酶、及纤维素酶中的β-葡萄糖苷酶组分,从而不再需要额外添加外源的β-葡萄糖苷酶,大大降低了工业应用纤维素酶的成本。
86.表2酶解总糖表
[0087][0088][0089]
其中,酶解总糖测定方法如下:
[0090]
①
100ml体系:称15g蒸汽爆破预处理后的玉米秸秆(195℃,15min)于250ml的摇瓶中,加入发酵纤维素酶液(45%),其余用水补足后,以玻璃棒搅匀,在摇床180rpm,50℃的条件下酶解24h;
[0091]
②
取出酶解液约2ml于玻璃试管中,放入沸水中煮沸5-10分钟,灭酶活性,再将试管中的酶解液倒入1.5ml的离心管中,离心取上清液,用纯净水稀释20倍。
[0092]
③
过膜,液相测葡萄糖、纤维二糖、木糖峰面积,并根据标曲公式计算计算葡萄糖(g/l)、纤维二糖(g/l)、木糖(g/l)浓度。酶解总糖浓度(g/l)计算如下:c
总糖
=c
葡萄糖
+c
纤维二糖
+c
木糖
。
[0093]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1