一种奥替溴胺的制备方法与流程
1.本发明属于药物制备技术领域,具体涉及一种奥替溴胺的制备方法。
背景技术:
2.奥替溴铵是一种抗毒蕈碱,其化学式为n,n-二乙基-n-甲基-2-[[4[[2-(辛基)苯甲酰]氨基]苯甲酰]氧基]乙烷铵溴化物,结构式如下:
[0003][0004]
它是一种白色粉末,熔点为30-133℃。临床上奥替溴铵是一种血小板活化因子的拮抗剂,可作为镇痛、抗炎、抑制子宫收缩和抗肿瘤剂。奥替溴铵也常用来解痉,作为斯巴敏的主要成分,适用于胃肠道痉挛和运动功能障碍,如肠易激综合征,胃炎,胃十二指肠炎,肠炎,食管病变等,也可用于内窥镜检查前准备,如食管-胃-十二指肠镜,结肠镜,直肠镜检查等。
[0005]
但是,对于奥替溴铵如何制备得到,现有技术中很少有文献记载。
技术实现要素:
[0006]
有鉴于此,本发明的目的是提供一种奥替溴胺的制备方法,该制备方法首先是由水杨酸甲酯和1-溴辛烷通过醚化反应制备得到中间体1、接着通过脱脂反应制备得到中间体2,再由中间体2和氯化亚砜通过酰氯化反应制备得到中间体3;然后由盐酸普鲁卡因通过酸碱中和制备得到中间体4;再由中间体3和中间体4通过成酰胺反应制备得到中间体5;最后中间体5和溴甲烷通过成盐反应制备得到奥替溴胺。本发明的制备方法工艺成熟,适合工业化生产,且产物纯度高(≥99.0%)、收率好(80%以上)。
[0007]
为了实现上述目的,本发明的技术方案具体如下:
[0008]
本发明提供一种奥替溴胺的制备方法,包括以下步骤:
[0009]
步骤1、醚化反应
[0010]
将甲醇钠甲醇溶液、水杨酸甲酯、1-溴辛烷加入第一反应釜中,第一反应釜的夹套内通入蒸汽,加热回流,反应7-9小时后,离心过滤,得副产品溴化钠,含中间体1的滤液进入第二反应釜减压蒸出甲醇;
[0011]
步骤2、脱酯反应
[0012]
向第二反应釜中加入naoh溶液,升温至90-100℃,脱酯;第二反应釜夹套内改通入冷冻盐水,降温至室温,用盐酸调ph=2~3;
[0013]
步骤3、二氯相蒸馏
[0014]
向第二反应釜中加入二氯甲烷,萃取,通过第二反应釜出料口的视镜观察,含中间体2的二氯相排入第三反应釜,在第三反应釜中进行二氯相蒸馏,回收二氯甲烷;
[0015]
步骤4、酰氯化反应
[0016]
用真空泵将二氯亚砜吸入第三反应釜,同时开启尾气吸收装置,第三反应釜夹套内通入蒸汽加热,升温到80-90℃回流4-6小时后,减压蒸馏回收二氯亚砜;第三反应釜加入二氯甲烷溶解,得到中间体3;
[0017]
步骤5、中间体4制备
[0018]
向第四反应釜中加入盐酸普鲁卡因,再滴加naoh溶液,将溶液的酸碱性调到ph=12-13,加入二氯甲烷萃取,通过第四反应釜出料口的视镜观察,含中间体4的二氯相排入第三反应釜;
[0019]
步骤6、中间体5制备
[0020]
将第三反应釜中的中间体3、4混合均匀后,滴加naoh溶液,第三反应釜夹套内通入冷冻盐水降温到0~5℃,反应12-14小时,得到中间体5,通过第三反应釜出料口的视镜观察,将含中间体5的二氯相排入第五反应釜;
[0021]
步骤7、奥替溴铵制备
[0022]
向第五反应釜中通入蒸汽,加热回流,回收二氯甲烷,向除去了二氯甲烷的中间体5中加入丙酮,搅拌全溶解,第五反应釜内改为通入冷冻盐水冷却,当温度降至15~20℃时,通入溴甲烷气体析出成品,离心机过滤,得奧替溴铵湿粗品,滤液进入第六反应釜经减压蒸馏回收丙酮;
[0023]
步骤8、奥替溴铵精制1
[0024]
将步骤7制备的奧替溴铵产品和乙醇加入到第七产品精制釜中,加热全部溶解,加入活性碳,搅拌30-40分钟,过滤,滤液进入第八反应釜,将乙醇减压浓缩尽,回收乙醇;
[0025]
步骤9、奥替溴铵精制2
[0026]
向第八反应釜中加丙酮,搅拌析晶、过滤,得固体成品,烘干,滤液进入第六反应釜经减压蒸馏回收丙酮。
[0027]
在上述技术方案中,优选的是,步骤1中甲醇钠甲醇溶液、水杨酸甲酯、1-溴辛烷的质量比为580kg:457kg:580kg,其中甲醇钠甲醇溶液中甲醇钠与甲醇的质量比为162.4kg:417.6kg。
[0028]
在上述技术方案中,优选的是,步骤1的反应时间是7小时。
[0029]
在上述技术方案中,优选的是,步骤2中naoh溶液的质量分数为8%。
[0030]
在上述技术方案中,优选的是,步骤2中升温至90℃。
[0031]
在上述技术方案中,优选的是,步骤4中升温到80℃回流4小时。
[0032]
在上述技术方案中,优选的是,步骤5和6中naoh溶液的质量分数为20%。
[0033]
在上述技术方案中,优选的是,步骤6中反应时间是12小时。
[0034]
在上述技术方案中,优选的是,步骤8中搅拌时间是30分钟。
[0035]
本发明的有益效果是:
[0036]
本发明提供的一种奥替溴胺的制备方法,首先是由水杨酸甲酯和1-溴辛烷通过醚化反应制备得到中间体1、接着通过脱脂反应制备得到中间体2,再由中间体2和氯化亚砜通过酰氯化反应制备得到中间体3;然后由盐酸普鲁卡因通过酸碱中和制备得到中间体4;再
由中间体3和中间体4通过成酰胺反应制备得到中间体5;最后中间体5和溴甲烷通过成盐反应制备得到奥替溴胺。本发明的制备方法工艺成熟,适合工业化生产,且产物纯度高(≥99.0%)、收率好(80%以上)。
附图说明
[0037]
下面结合附图和具体实施方式对本发明作进一步详细说明。
[0038]
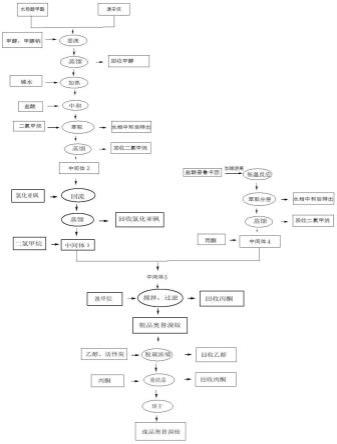
图1为本发明的奥替溴胺的制备方法的工艺流程图。
具体实施方式
[0039]
实施例1
[0040]
结合图1具体说明本发明提供一种奥替溴胺的制备方法,包括以下步骤:
[0041]
步骤1、醚化反应
[0042]
将580kg甲醇钠甲醇溶液(其中甲醇钠162.4kg,甲醇417.6kg)、457kg水杨酸甲酯、580kg 1-溴辛烷加入第一反应釜中,第一反应釜的夹套内通入蒸汽(0.6mpa,164℃),加热回流,反应7小时后,离心过滤,得副产品溴化钠,含中间体1的滤液进入第二反应釜减压蒸出甲醇(套用);
[0043]
中间体1的合成反应式如下:
[0044][0045]
中间体1经过气相色谱-质谱(gc-ms)表征:产物分子量为:264.34,理论分子量为264.37,实测值与理论值相符。
[0046]
步骤2、脱酯反应
[0047]
向第二反应釜中加入质量分数为8%的naoh溶液80l,升温至90℃,脱酯;第二反应釜夹套内改通入氯化钙冷冻盐水,降温至室温,用盐酸调ph=2;
[0048]
步骤3、二氯相蒸馏
[0049]
向第二反应釜中加入二氯甲烷,萃取,通过第二反应釜出料口的视镜观察,含中间体2的二氯相排入第三反应釜,在第三反应釜中进行二氯相蒸馏,回收二氯甲烷(套用);
[0050]
步骤4、酰氯化反应
[0051]
用真空泵将139.6kg二氯亚砜吸入第三反应釜,同时开启尾气吸收装置,第三反应釜夹套内通入蒸汽(0.6mpa,164℃)加热,升温到80℃回流4小时后,减压蒸馏回收二氯亚砜(套用);第三反应釜加入二氯甲烷溶解,得到中间体3;
[0052]
中间体2和3的合成反应式如下:
[0053][0054]
中间体2经过气相色谱-质谱(gc-ms)表征:产物分子量为:250.33,理论分子量为250.34,实测值与理论值相符。
[0055]
中间体3经过气相色谱-质谱(gc-ms)表征:产物分子量为:268.74,理论分子量为268.78,实测值与理论值相符。
[0056]
步骤5、中间体4制备
[0057]
向第四反应釜中加入660kg盐酸普鲁卡因,再滴加质量分数为20%naoh溶液,将溶液的酸碱性调到ph=12,加入二氯甲烷萃取,通过第四反应釜出料口的视镜观察,含中间体4的二氯相排入第三反应釜;
[0058]
中间体4的合成反应式如下:
[0059][0060]
中间体4经过气相色谱-质谱(gc-ms)表征:产物分子量为:236.30,理论分子量为236.32,实测值与理论值相符。
[0061]
步骤6、中间体5制备
[0062]
将第三反应釜中的中间体3、4混合均匀后,滴加质量分数为20%的naoh溶液510l,第三反应釜夹套内通入氯化钙冷冻盐水降温到0℃,反应12小时,得到中间体5,通过第三反应釜出料口的视镜观察,将含中间体5的二氯相排入第五反应釜;
[0063]
中间体5的合成反应式如下:
[0064][0065][0066]
中间体5经过气相色谱-质谱(gc-ms)表征:产物分子量为:468.61,理论分子量为468.64,实测值与理论值相符。
[0067]
步骤7、奥替溴铵制备
[0068]
向第五反应釜中通入蒸汽(0.6mpa,164℃),加热回流,回收二氯甲烷(套用),向除去了二氯甲烷的中间体5中加入丙酮,搅拌全溶解,第五反应釜内改为通入氯化钙冷冻盐水冷却,当温度降至15℃时,通入235kg溴甲烷气体析出成品,离心机过滤,得奧替溴铵湿粗品,滤液进入第六反应釜经减压蒸馏回收丙酮(套用);
[0069][0070]
步骤8、奥替溴铵精制1
[0071]
将步骤7制备的奧替溴铵产品和乙醇加入到第七产品精制釜中,加热全部溶解,加入6kg活性碳,搅拌30分钟,过滤,滤液进入第八反应釜,将乙醇减压浓缩尽,回收乙醇(套用);
[0072]
步骤9、奥替溴铵精制2
[0073]
向第八反应釜中加丙酮,搅拌析晶、过滤,得固体成品,烘干,滤液进入第六反应釜经减压蒸馏回收丙酮(套用)。
[0074]
所得成品奥替溴铵为白色粉末,熔点:130-133℃。产物经过气相色谱-质谱(gc-ms)表征:产物分子量为:563.52,理论分子量为563.57,实测值与理论值相符;通过气相色谱进行(gs)测定,纯度≥99.0%;收率达到85%。
[0075]
实施例2
[0076]
结合图1具体说明本发明提供一种奥替溴胺的制备方法,包括以下步骤:
[0077]
步骤1、醚化反应
[0078]
将580kg甲醇钠甲醇溶液(其中甲醇钠162.4kg,甲醇417.6kg)、457kg水杨酸甲酯、580kg 1-溴辛烷加入第一反应釜中,第一反应釜的夹套内通入蒸汽(0.6mpa,164℃),加热回流,反应7小时后,离心过滤,得副产品溴化钠,含中间体1的滤液进入第二反应釜减压蒸出甲醇(套用);
[0079]
中间体1的合成反应式如下:
[0080][0081]
中间体1经过气相色谱-质谱(gc-ms)表征:产物分子量为:264.36,理论分子量为264.37,实测值与理论值相符。
[0082]
步骤2、脱酯反应
[0083]
向第二反应釜中加入质量分数为8%的naoh溶液80l,升温至90℃,脱酯;第二反应釜夹套内改通入氯化钙冷冻盐水,降温至室温,用盐酸调ph=2;
[0084]
步骤3、二氯相蒸馏
[0085]
向第二反应釜中加入二氯甲烷,萃取,通过第二反应釜出料口的视镜观察,含中间
体2的二氯相排入第三反应釜,在第三反应釜中进行二氯相蒸馏,回收二氯甲烷(套用);
[0086]
步骤4、酰氯化反应
[0087]
用真空泵将139.6kg二氯亚砜吸入第三反应釜,同时开启尾气吸收装置,第三反应釜夹套内通入蒸汽(0.6mpa,164℃)加热,升温到80℃回流4小时后,减压蒸馏回收二氯亚砜(套用);第三反应釜加入二氯甲烷溶解,得到中间体3;
[0088]
中间体2和3的合成反应式如下:
[0089][0090]
中间体2经过气相色谱-质谱(gc-ms)表征:产物分子量为:250.29,理论分子量为250.34,实测值与理论值相符。
[0091]
中间体3经过气相色谱-质谱(gc-ms)表征:产物分子量为:268.72,理论分子量为268.78,实测值与理论值相符。
[0092]
步骤5、中间体4制备
[0093]
向第四反应釜中加入660kg盐酸普鲁卡因,再滴加质量分数为20%naoh溶液510l,将溶液的酸碱性调到ph=12,加入二氯甲烷萃取,通过第四反应釜出料口的视镜观察,含中间体4的二氯相排入第三反应釜;
[0094]
中间体4的合成反应式如下:
[0095][0096]
中间体4经过气相色谱-质谱(gc-ms)表征:产物分子量为:236.32,理论分子量为236.32,实测值与理论值相符。
[0097]
步骤6、中间体5制备
[0098]
将第三反应釜中的中间体3、4混合均匀后,滴加质量分数为20%的naoh溶液525l,第三反应釜夹套内通入氯化钙冷冻盐水降温到0℃,反应12小时,得到中间体5,通过第三反应釜出料口的视镜观察,将含中间体5的二氯相排入第五反应釜;
[0099]
中间体5的合成反应式如下:
[0100][0101]
中间体5经过气相色谱-质谱(gc-ms)表征:产物分子量为:468.63,理论分子量为468.64,实测值与理论值相符。
[0102]
步骤7、奥替溴铵制备
[0103]
向第五反应釜中通入蒸汽(0.6mpa,164℃),加热回流,回收二氯甲烷(套用),向除去了二氯甲烷的中间体5中加入丙酮,搅拌全溶解,第五反应釜内改为通入氯化钙冷冻盐水冷却,当温度降至15℃时,通入235kg溴甲烷气体析出成品,离心机过滤,得奧替溴铵湿粗品,滤液进入第六反应釜经减压蒸馏回收丙酮(套用);
[0104][0105]
步骤8、奥替溴铵精制1
[0106]
将步骤7制备的奧替溴铵产品和乙醇加入到第七产品精制釜中,加热全部溶解,加入6kg活性碳,搅拌30分钟,过滤,滤液进入第八反应釜,将乙醇减压浓缩尽,回收乙醇(套用);
[0107]
步骤9、奥替溴铵精制2
[0108]
向第八反应釜中加丙酮,搅拌析晶、过滤,得固体成品,烘干,滤液进入第六反应釜经减压蒸馏回收丙酮(套用)。
[0109]
所得成品奥替溴铵为白色粉末,熔点:130-133℃。产物经过气相色谱-质谱(gc-ms)表征:产物分子量为:563.55,理论分子量为563.57,实测值与理论值相符;通过气相色谱进行(gs)测定,纯度≥99.0%;收率达到81%。
[0110]
本发明提供的一种奥替溴胺的制备方法,该制备方法首先是由水杨酸甲酯和1-溴辛烷通过醚化、脱脂反应制备得到中间体1,再由中间体1和氯化亚砜通过酰氯化反应制备得到中间体2;然后由盐酸普鲁卡因通过酸碱中和制备得到中间体3;再由中间体2和中间体3通过成酰胺反应制备得到中间体4;最后中间体4和溴甲烷通过成盐反应制备得到奥替溴胺。本发明的制备方法工艺成熟,适合工业化生产,且产物纯度高(≥99.0%)、收率好(80%以上)。
[0111]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对
于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1