一锅法制备维兰特罗中间体的方法与流程
1.本发明属于化学合成领域,更具体地说,涉及一锅法制备维兰特罗中间体的方法。
技术背景
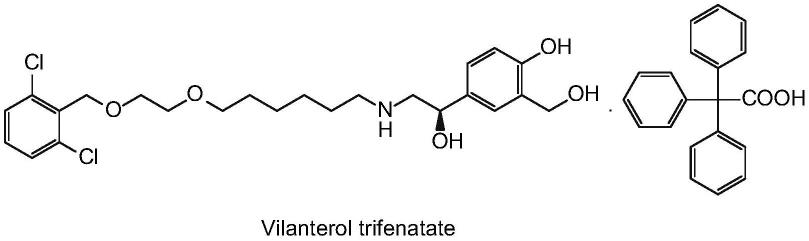
2.三苯乙酸维兰特罗是一种长效β2受体激动剂,具有长效支气管扩张作用,适用于慢性阻塞性肺病(copd)的长期维持治疗。三苯基乙酸-4-{(1r)-2-[(6-{2-[(2,6-二氯苄基)氧基]乙氧基}己基)氨基]-1-羟乙基}-2-(羟甲基)苯酚,其结构式如下:
[0003][0004]
葛兰素史克公司在国际专利申请公开号wo2003024439a1中使式i所示化合物在三甲基硅醇钾存在下进行开环反应,得到式ii所示化合物后,再利用盐酸溶液进行脱保护基,生成式iii所示化合物,式iii所示化合物与三苯基乙酸反应生成盐,得到维兰特罗三苯乙酸盐,其合成路线如下所示:
[0005][0006]
该方法获得的中间体ii纯度较低,杂质较多,不适合工业化生产。
[0007]
中国专利申请公开号cn104744271a中提出将式iv所示化合物和式v所示化合物进行脱水反应生成西弗碱,再还原得到式ii所示化合物,然后在酸性条件下去除保护基,形成式iii所示化合物。式iii所示化合物与三苯乙酸成盐,得到维兰特罗三苯乙酸盐,合成路线如下所示:
[0008]
[0009]
该方法使用了易制爆的硼氢化钠,危险性较高且整个工艺收率不高。
[0010]
中国专利申请公开号cn113121492a公开了上述式ii所示化合物的盐的制备方法,反应式如下:
[0011][0012]
其中x表示三苯乙酸、琥珀酸、柠檬酸、水杨酸、肉桂酸或苯甲酸。
[0013]
具体过程为:将式ii所示化合物与三苯乙酸、琥珀酸、柠檬酸、水杨酸、肉桂酸或苯甲酸,在乙酸乙酯或c
1-c4的醇类溶剂中形成式所示i化合物(式ii化合物的盐)。提到了形成盐的步骤的收率为87.1~95.7%,这些盐的纯度为99.0%~99.6%,异构体杂质含量未提及。其中式ii化合物按照wo2014041565a2公开的方法制备,具体为:将式i所示化合物在thf中在三甲基硅醇钾存在下,开环生成式ii所示化合物,反应完成后,反应液利用5%磷酸钠溶液处理,并用乙酸乙酯萃取,浓缩有机相得到式ii所示化合物的粗品,然后进行柱色谱纯化,得到纯度比较高的式ii化合物。反应式如下:
[0014][0015]
专利申请公开号cn111807973a中公开了上述式ii所示化合物的丁二酸盐(即,ii-1化合物)的制备方法,反应式如下:
[0016][0017]
具体为:将式i所示化合物在四氢呋喃中,在三甲基硅醇钾存在下加热回流,tlc检测反应完全。反应液冷却至室温,加入h3po4/kh2po4缓冲液,调节ph至8,室温搅拌10分钟,用乙酸乙酯萃取3遍,合并有机相,水洗2遍,在减压下,将有机相浓缩,制得式ii所示化合物粗品,为淡黄色油状物,该淡黄色油状物在乙醇、异丙醇、乙酸乙酯中与丁二酸成盐,过滤得式ii所示化合物的丁二酸盐,收率为76.1~70.6%,hplc纯度为99.5~99.7%,异构体杂质0.07%~0.09%。
[0018]
也就是说,按照中国专利申请公开号cn113121492a和cn111807973a公开的方法制备式ii所示化合物的盐的过程中,均需要将含式ii所示化合物的反应液进行处理后,用大量的溶剂进行萃取,浓缩得到式ii所示化合物粗品,然后再与酸反应,制备相应的盐。处理含式ii所示化合物的反应液,需要利用酸性溶液,且萃取需要使用大量的溶剂,并产生大量的废液,环境污染大。
[0019]
因此,从产品的纯度、收率、制备过程等方面考虑,有必要开发一种简单高效的式
ii所示化合物及其盐的制备方法,简化生产步骤及后处理工序,减少溶剂使用量和三废排放量,从而实现降低生产成本的目的,使得三苯乙酸维兰特罗更加适合工业规模化生产。
技术实现要素:
[0020]
针对现有技术的不足,本发明一方面的目的是提供一种一锅法制备维兰特罗中间体,式ii所示化合物的富马酸盐的方法,该方法具有去杂效果好、工艺稳定可靠、操作简便,产品纯度和收率高的优点。
[0021]
本发明的一锅法制备式ii所示化合物的富马酸盐的方法包括以下步骤:
[0022]
(1)式i所示化合物在有机溶剂中,在碱存在下,开环生成式ii所示化合物,反应完成后,向反应液中加入富马酸调节其ph为6~7,析出式ii所示化合物的富马酸盐,反应式如下:
[0023][0024]
所述碱可选自甲醇钠、乙醇钠、叔丁醇钾、叔丁醇钠、氢氧化钠、氢氧化钾、氢氧化锂、三甲基硅醇钾,或其组合。
[0025]
在本发明的一些具体实施例中,所述碱选自氢氧化钾、乙醇钠,或其组合。
[0026]
所述有机溶剂可选自甲醇、乙醇、异丙醇、正丁醇、甲苯、甲基叔丁基醚、乙酸乙酯、乙酸异丙酯、乙酸丁酯、四氢呋喃、2-甲基四氢呋喃,或其组合。
[0027]
在本发明的一些具体实施例中,所述有机溶剂选自异丙醇、乙醇,或其组合。
[0028]
式i所示化合物与碱的摩尔量比为1:1~4.0。
[0029]
在本发明的一些具体实施例中,式i所示化合物与碱的摩尔量比为1:1.5~2.5。
[0030]
有机溶剂与式i所示化合物的体积重量比为3~10l/g,更优选4~6l/g。
[0031]
所述有机溶剂与式i所示化合物的体积重量比为4.0~6.0:1。
[0032]
步骤(1)中反应温度为50~100℃。
[0033]
在本发明的一些具体实施例中,步骤(1)中反应温度为55~75℃。
[0034]
式i所示化合物与富马酸的摩尔量比为1:1.5~2.8。
[0035]
在本发明的一些具体实施例中,式i所示化合物与富马酸的摩尔量比为1:1.6。
[0036]
步骤(1)中加入富马酸调节反应液的ph过程中,保持反应液的温度为0~40℃。
[0037]
本发明另一方面提供了式ii所示化合物的富马酸盐的晶型a,所述晶型a的x-射线粉末衍射在下列衍射角2θ处具有特征峰:4.6
±
0.2
°
、9.2
±
0.2
°
、14.0
±
0.2
°
和23.4
±
0.2
°
。
[0038]
式ii所示化合物的富马酸盐的晶型a的x射线粉末衍射还在下列一个或多个衍射
角2θ处具有特征峰:
[0039]
18.4
±
0.2
°
、19.5
±
0.2
°
、20.6
±
0.2
°
、21.8
±
0.2
°
、25.5
±
0.2
°
、28.1
±
0.2
°
。
[0040]
式ii所示化合物的富马酸盐的晶型a的差示扫描量热(dsc)谱图在140℃~146℃具有吸热峰。
[0041]
本发明再一方面提供了一种维兰特罗的制备方法,该制备方法包括以下步骤:
[0042]
(1)将上述式ii所示化合物的富马酸盐溶于溶剂中,形成溶液,向所述溶液中加入盐酸,在0~40℃保持20~30小时,反应液调节为ph=9~10,利用有机溶剂萃取,分层得到含有ii所示化合物的有机相;
[0043]
其中所述盐酸的浓度为0.1mol/l~1.0mol/l,且加入盐酸的体积是所述溶液体积的0.5~3倍,
[0044]
(2)向所述有机相中加入三苯乙酸成盐,搅拌析晶,过滤得到三苯乙酸维兰特罗。
[0045]
步骤(1)中,所述溶液中加入盐酸过程中保持溶液的温度为0~15℃。在本发明的一些具体实施例中,步骤(1)中,所述溶液中加入盐酸过程中保持溶液的温度为0~10℃。
[0046]
溶解式ii所示化合物的富马酸盐的溶剂可选自甲醇、乙醇、异丙醇、正丁醇、甲苯、甲基叔丁基醚、乙酸乙酯、乙酸异丙酯、乙酸丁酯、四氢呋喃、2-甲基四氢呋喃,或其组合。在本发明的一些具体实施例中,溶解式ii所示化合物的富马酸盐的溶剂选自异丙醇,乙醇,或其组合。
[0047]
用于萃取的有机溶剂可选自乙酸乙酯、甲基叔丁基醚、异丙醚、乙酸异丙酯、乙酸丁酯。在本发明的一些具体实施例中,用于萃取的有机溶剂选自乙酸乙酯。
[0048]
在本发明的一些具体实施例中,所述盐酸的浓度为0.4mol/l~0.6mol/l,且加入盐酸的体积是所述溶液体积的1~1.5倍。
[0049]
调节反应液ph值所用的碱选自碳酸钠、碳酸钾、氢氧化钠、氢氧化钾,或其组合。
[0050]
调节反应液ph值所用的碱优选碱的水溶液。例如,质量浓度为10~40%的碱溶液。
[0051]
利用碱的水溶液调节反应液ph值过程中优选保持体系温度为0~15℃。
[0052]
步骤(2)中三苯乙酸与式ii所示化合物的富马酸盐的摩尔比为2~8:1。在本发明的一些具体实施例中,三苯乙酸与式ii所示化合物的富马酸盐的摩尔比为4~6:1。
[0053]
与现有技术相比,本发明达到了以下优势效果:
[0054]
(1)本发明的一锅法制备式ii所示化合物的富马酸盐的方法反应结束后使用富马酸直接调节反应液的ph为6~7,搅拌即可析出式ii所示化合物的富马酸盐的晶体,析出的晶体无需任何处理即可达到纯度为99.7~100.0%,异构体含量为0.01~0.03%,且产率高。
[0055]
(2)本发明的一锅法制备式ii所示化合物的富马酸盐的方法避免了现有技术提到的步骤:从式i所示化合物制备式ii所示化合物的反应完成后,使用磷酸二氢钾溶液调节反应液ph,淬灭反应,多次萃取分层工序,以及利用柱层析分离纯化得到纯度相对较高的式ii所示化合物。
[0056]
(3)本发明的一锅法制备式ii所示化合物的富马酸盐的方法中,富马酸既作为淬灭试剂,又作为成盐试剂,与上述现有技术中制备式ii所示化合物的其他盐相比,大大减少了制备过程中溶剂的使用量以及三废排放量,节约了成本。而且该方法的重复性好,成盐产物纯度高、产率高、稳定性好,便于贮存,适合工业化生产。
附图说明
[0057]
图1为本发明实施例1制备的式ii所示化合物的富马酸盐的hplc检测图谱;
[0058]
图2为本发明实施例1制备的式ii所示化合物的富马酸盐的x-射线粉末衍射谱图;
[0059]
图3为本发明实施例1制备的式ii所示化合物的富马酸盐的差示扫描量热谱图;
[0060]
图4为本发明对比例1制备的式ii所示化合物的三苯乙酸盐的hplc检测图谱;
[0061]
图5为本发明对比例2制备的式ii所示化合物的富马酸盐的hplc检测图谱;
[0062]
图6为本发明对比例3制备的式ii所示化合物的丁二酸盐的hplc检测图谱。
具体实施方式
[0063]
针对现有技术存在的缺点,本技术发明人经过深入的研究发现,利用式i所示化合物为原料,在碱催化下,进行开环得到式ii所示化合物,反应完成后,直接利用富马酸盐调节反应液的ph至6~7,即可析出纯度好、产率高的式ii所示化合物的富马酸盐。在此基础上完成了本发明。
[0064]
一锅法制备式ii所示化合物的富马酸盐
[0065]
(1)式i所示化合物在有机溶剂中,在碱存在下,开环生成式ii所示化合物,反应完成后,向反应液中加入富马酸调节其ph至6~7,析出式ii所示化合物的富马酸盐(式iii所示化合物),反应式如下:
[0066][0067]
反应所用的碱包括但不局限于甲醇钠、乙醇钠、叔丁醇钾、叔丁醇钠、氢氧化钠、氢氧化钾、氢氧化锂、三甲基硅醇钾。在本发明的一个优选实施例中,所述碱为氢氧化钾或乙醇钠。反应所用的有机溶剂包括但不局限于甲醇、乙醇、异丙醇、正丁醇、甲苯、甲基叔丁基醚、乙酸乙酯、乙酸异丙酯、乙酸丁酯、四氢呋喃、2-甲基四氢呋喃。在本发明的一些优选实施例中,所述有机溶剂为异丙醇或乙醇。式i所示化合物与碱的摩尔量比为1:1~4.0。在本发明的一些优选实施例中,式i所示化合物与碱的摩尔量比为1:1.5~2.5,例如1:1.5、1:2.0、1:2.5。有机溶剂与式i所示化合物的体积重量(ml/g)比为3~10:1。在本发明的一些优选实施例中,有机溶剂与式i所示化合物的体积重量(ml/g)比为4.0~6.0:1,例如4.0:1、4.5:1、5.0:1、5.5:1、6.0:1。步骤(1)中反应温度为50~80℃。在本发明的一些优选实施例中,步骤(1)中反应温度为55~75℃,例如56℃、60℃、65℃、70℃、75℃。式i所示化合物与富马酸的摩尔量比为1:1.5~2.8。在本发明的一些优选实施例中,式i所示化合物与富马酸的摩尔量比为1:1.5~2.5,例如1:1.5、1:1.6、1:1.8、1:2.0、1:2.5。加入富马酸调节反应液的ph过程中,保持反应液的温度为0~40℃。
[0068]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验过程,通常按照常规条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
[0069]
下列实施例中所用hplc分析测试仪器为agilent 1260,色谱柱为zorbax sb c18 150*4.6mm,3.5um,检测器为uv检测器,波长为220nm,流动相a为0.1%磷酸水溶液,流动相b为乙腈,洗脱程序是0-5min为90%流动相a和10%流动相b,5-40min为20%流动相a和80%流动相b,40-50min为90%流动相a和10%流动相b。
[0070]
x射线粉末衍射检测所用仪器为miniflex 600-c,x射线光源为cu,kα。
[0071]
差示扫描量热(dsc)检测所用仪器为shimadzu差示扫描量热仪dsc-60,扫描速率为5℃/min,保护气体为氮气。
[0072]
下列实施例中,式ⅰ所示化合物是按照专利申请号cn103288793a制备得到。
[0073]
实施例1
[0074]
将11.05g式ⅰ所示化合物(纯度90.69%,异构体含量0.83%),2.24g氢氧化钾溶于55.25ml异丙醇中,升温至70℃,反应6h后反应结束,降至室温,然后加入3.71g富马酸调节溶液ph至6~7,搅拌1h后析出固体,过滤,烘干后得到11.05g式ii所示化合物的富马酸盐,为白色或类白色固体,其hplc纯度为99.88%(参见图1),无异构体,摩尔收率为99.22%(折算原料与产物的纯度后)。
[0075]
将实施例1得到的白色固体进行x射线粉末衍射测定,得到如图2所示的图谱。从该图可以看出,特征峰位置为2θ(
±
0.2
°
)=4.6
°
、9.2
°
、14.0
°
、18.4
°
、19.5
°
、20.6
°
、21.8
°
、23.4
°
、25.5
°
、28.1
°
。
[0076]
将实施例1得到的白色固体进行差示扫描量热分析,得到如图3所示的图谱。从该图可以看出,差示扫描量热(dsc)谱图在约140℃至约146℃之间具有吸热峰。
[0077]
实施例2
[0078]
同实施例1的操作一样(反应条件、溶剂、反应物及其用量等均同实施例1一样),不同之处为将2.24g氢氧化钾替换为1.12g氢氧化钾,最终得到10.50g式ii所示化合物的富马酸盐,hplc纯度为99.72%,异构体含量为0.02%,摩尔收率为94.11%(折算原料与产物的纯度后)。
[0079]
实施例3
[0080]
同实施例1的操作一样(反应条件、碱催化剂、反应物及其用量均同实施例1一样),不同之处为将55.25ml异丙醇替换为88.4ml异丙醇,最终得到10.06g式ii所示化合物的富马酸盐,hplc纯度为99.79%,异构体含量为0.02%,摩尔收率为90.21%(折算原料与产物的纯度后)。
[0081]
实施例4
[0082]
同实施例1的操作一样(溶剂、碱催化剂、反应物及其用量等均同实施例1一样),不同之处为将升温至70℃替换为升温至50℃,最终得到9.83g式ii所示化合物的富马酸盐,hplc纯度为99.69%,异构体含量为0.03%,摩尔收率为88.14%(折算原料与产物的纯度后)。
[0083]
实施例5
[0084]
同实施例1的操作一样(反应条件、溶剂、碱催化剂及其用量等均同实施例1一样),
不同之处为将3.71g富马酸替换为5.11g富马酸,最终得到10.94g式ii所示化合物的富马酸盐,hplc纯度为99.83%,异构体含量为0.01%,摩尔收率为98.18%(折算原料与产物的纯度后)。
[0085]
实施例6
[0086]
同实施例1的操作一样(反应条件、碱催化剂、反应物及其用量等均同实施例1一样),不同之处为将55.25ml异丙醇替换为55.25ml乙醇,最终得到10.83g式ii所示化合物的富马酸盐,hplc纯度为99.82%,异构体含量为0.01%,摩尔收率为97.18%(折算原料与产物的纯度后)。
[0087]
实施例7
[0088]
同实施例1的操作一样(反应条件、碱催化剂、反应物及其用量等均同实施例1一样),不同之处为将55.25ml异丙醇替换为55.25ml 2-甲基四氢呋喃,最终得到10.59g式ii所示化合物的富马酸盐,hplc纯度为99.72%,异构体含量为0.02%,摩尔收率为94.90%(折算原料与产物的纯度后)。
[0089]
实施例8
[0090]
同实施例1的操作一样(反应条件、溶剂、反应物及其用量等均同实施例1一样),不同之处为将2.24g氢氧化钾替换为2.72g乙醇钠,最终得到10.72g式ii所示化合物的富马酸盐,hplc纯度为99.79%,异构体含量为0.02%,摩尔收率为96.16%(折算原料与产物的纯度后)。
[0091]
实施例9
[0092]
同实施例1的操作一样(反应条件、溶剂、反应物及其用量等均同实施例1一样),不同之处为将2.24g氢氧化钾替换为4.49g叔丁醇钾,最终得到10.61g式ii所示化合物的富马酸盐,hplc纯度为99.81%,异构体含量为0.01%,摩尔收率为95.18%(折算原料与产物的纯度后)。
[0093]
实施例10
[0094]
同实施例1的操作一样(反应条件、溶剂、反应物及其用量等均同实施例1一样),不同之处为将2.24g氢氧化钾替换为5.13g三甲基硅醇钾,最终得到10.39g式ii所示化合物的富马酸盐,hplc纯度为99.68%,异构体含量为0.03%,摩尔收率为93.08%(折算原料与产物的纯度后)。
[0095]
实施例11
[0096]
将实施例1获得的式ii所示化合物的富马酸盐10g,加入100ml乙醇,降温至0~10℃后,加入200ml 0.5n稀盐酸溶液,保温反应24h后,加入500ml乙酸乙酯,控温0~10℃下缓慢滴加20%碳酸钠水溶液,调节ph=9~10,分层后有机层再用40ml水洗涤,分层,然后加入5g三苯乙酸,搅拌3h后,过滤,获得12g三苯乙酸维兰特罗,hplc纯度99.60%。
[0097]
对比例1
[0098]
将11.05g式ⅰ所示化合物(纯度90.69%,异构体含量0.83%),2.24g氢氧化钾溶于55.25ml异丙醇,升温至70℃,反应6h后反应结束,降至室温,然后加入12.69g三苯乙酸调节溶液ph至6~7,搅拌1h后析出固体,过滤,烘干后得到11.78g式ii所示化合物的三苯乙酸盐(hplc纯度99.33%,异构体含量0.16%),摩尔收率79.19%(折算原料与产物的纯度后)。
[0099]
对比例2
[0100]
将11.05g式ⅰ所示化合物(纯度90.69%,异构体含量0.83%),2.24g氢氧化钾溶于55.25ml氯仿中,升温至70℃,反应6h后反应结束,降至室温,然后加入3.71g富马酸调节溶液ph至6~7,搅拌1h后析出固体,过滤,烘干,由于反应不完全,导致最终只得到7.18g式ii所示化合物的富马酸盐,hplc纯度99.52%,无异构体,摩尔收率64.26%(折算原料与产物的纯度后)。
[0101]
对比例3
[0102]
将11.05g式ⅰ所示化合物(纯度90.69%,异构体含量0.83%),2.24g氢氧化钾溶于55.25ml异丙醇中,升温至70℃,反应6h后反应结束,降至室温,然后加入12.69g丁二酸调节溶液ph至6~7,搅拌1h后析出固体,过滤,烘干后得到10.16g式ii所示化合物的丁二酸盐(即,上述现有技术中提到的化合物ii-1),hplc纯度99.33%,异构体含量0.14%,摩尔收率86.31%(折算原料与产物的纯度后)。
[0103]
从以上实施例1~10和对比例1和3的对比可以看出,与式ii所示化合物的三苯乙酸盐和丁二酸盐相比,式ii所示化合物的富马酸盐在相同的操作条件下纯度更高、异构体含量更少,摩尔收率更高,更适合工业化生产。
[0104]
从以上实施例1~10和对比例2的对比可以看出,醇类试剂比氯仿更适合式ⅰ所示化合物的开环反应,使用前者为溶剂时反应效率更高。
[0105]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1