一种多肽固相合成方法与流程
1.本发明涉及一种多肽固相合成方法。
背景技术:
2.多肽通常是指10~100个氨基酸通过肽键连接而成的化合物,其连接方式与蛋白质相同,它是生物体内天然存在的生物活性物质,从发现至今已有超过百年的历史。在生物体内发现的多肽已达上万种,其生物活性广泛而重要,能够广泛作用于内分泌系统、免疫系统、消化系统、心血管系统、血液系统、肌肉骨骼系统等。多肽作为药物应用也已超过70年,凭借其毒性低、特异性高、分子量小等独特优势而受到越来越多的关注,截止目前全球已有超过100个多肽类药物获批,这其中不乏如格拉替雷、奥曲肽、利拉鲁肽、度拉糖肽和索马鲁肽等年销售额在数十亿美元的重磅品种。
3.目前市场上的多肽类产品主要来源于动物组织提取,基因重组表达和化学合成三种生产方式。来自动物组织提取的多肽类药物将逐步被淘汰,而以dna重组技术为主导的基因工程法在一定程度上解决了多肽生产难的问题,但有些序列生产周期长、表达效率低,拿到的产物甚至难以进行纯化,另外还有部分复杂多肽分子尤其是含有非天然氨基酸以及修饰的多肽根本无法通过生物表达来实现。化学合成方式显然是最直接的多肽类药物合成方法,可以方便快捷地对多肽的结构进行改造,如引入非天然氨基酸以提高稳定性、进行化学修饰以调节其亲疏水性以及与某些靶标的亲和性等,从而提升多肽分子的成药性。化学合成法主要有液相合成法和固相合成法,前者一般用于较短序列的多肽的合成,而目前多肽药物的结构复杂度越来越高,其合成普遍采用的是多肽固相合成技术。
4.多肽固相合成技术是1963年由美国化学家r. bruce merrifield教授发明的,在多肽化学上具有里程碑意义。其基本原理如图1所示,是以连接于不溶性载体的第一个氨基酸为起点,经活化、偶合、脱临时性保护基团的循环过程,将保护氨基酸(半永久性保护基团保护活性侧链)逐一“组装”形成目标多肽分子,最后将多肽从不溶性载体上裂解下来并同时去除半永久性保护基团后,得到多肽产物。这种方法的显著优点为:简化并加速了多步骤的合成,可避免因手工操作和物料重复转移而产生的损失;能够通过快速的抽滤、洗涤完成未反应物的分离,避免了液相肽合成中冗长的中间体分离纯化步骤(重结晶或硅胶柱纯化)带来的大量的损失;通过使用过量反应物,促使个别困难反应进行完全,以实现最终产物的高产率;固相载体的使用可以缓解肽链的自聚集,有利于合成反应的顺利进行;可以在一个反应容器中进行所有的反应,便于自动化操作等。尤其值得一提的是,固相合成法通用性高,初期工艺开发时间短,这些特点都使其非常适用于多肽药物的开发。随着这种固相合成方式的开发和优化,大量具有重要生物活性的多肽、激素及一些蛋白质在短时间被一一合成,极大促进了生命科学研究的发展,为此merrifield教授于1984年荣获诺贝尔化学奖。
5.固相合成中最早是用苄氧羰基(z)作为临时性保护基团(α氨基的保护基),它的脱除需要较强的酸解条件,故后来采用在三氟乙酸(tfa)条件下即可脱除的叔丁氧羰基(boc)保护基,脱除条件相对温和且脱除过程中不易发生消旋。但此法仍不适用含有色氨酸等对
酸不稳定的肽类的合成,并且最终的裂解多采用腐蚀性较强的氢氟酸法或三氟甲磺酸法,对设备要求非常苛刻。
6.后来,carpino等人开发了芴甲氧羰基(fmoc)用作多肽固相合成中氨基酸原料的α氨基保护基,它能用哌啶等仲胺分子脱除,但对酸比较稳定故可与叔丁基(tbu)、三苯甲基(trt)等半永久性保护基团搭配使用,如此一来最终的裂解在tfa中就能实现,过程比较温和,对设备的要求也低得多。近年来,基于fmoc/tbu法的多肽固相合成得到了广泛的应用,其具体合成由下列四个步骤组成:1)去保护:需用一种碱性溶剂(如哌啶)处理树脂,去除上面氨基的保护基团fmoc以释放α氨基;2)活化和偶合:下一个待偶合的氨基酸的羧基被活化剂活化后,与释放出的α氨基进行偶合反应,从而形成肽键;3)将步骤1)、2)循环进行直到合成完成;4)裂解:将多肽从树脂上裂解下来,同时氨基酸侧链半永久保护基也被脱除,最后将裂解液加入至乙醚或甲基叔丁基醚中进行析晶,从而得到多肽产物。
7.基于fmoc/tbu法的多肽固相合成技术有众多优点,但由于其是一个连续生产过程,每一个步骤中由于副反应如断裂、缺失、异构化、氧化、还原、水解等产生的错误序列等都将累积到最终的多肽产物中,这就给后续的多肽纯化工艺(主要是高效液相色谱纯化)带来了巨大挑战,尤其是在制备较长的多肽序列或者易于形成二级结构的多肽序列时,问题将变得非常严重,如粗肽(即裂解后的多肽产物)纯度较低、合成收率不理想、影响纯化的杂质较多等。这实际上也是限制多肽固相合成技术应用到更长的多肽序列乃至蛋白质合成领域中的主要原因。因此,合成高纯度的多肽产物无疑是提高固相法多肽生产工艺效率(合成效率及纯化效率)的最重要的方向之一。
技术实现要素:
8.本发明的目的是提供一种在多肽的固相合成中、尤其在合成一些序列较长、易形成二级结构的多肽序列时,有效提高多肽产物纯度的多肽固相合成方法。
9.本发明采用如下技术方案:一种多肽固相合成方法,其包括如下步骤:(a)脱除固相树脂上的氨基酸临时保护基团;(b)依次使用酸性溶液和碱性溶液对所述固相树脂进行浸泡或冲洗;(c)通过逐一缩合的方式按照目标序列依次进行氨基酸的偶合,以合成具有目标序列的多肽;(d)将所述具有目标序列的多肽从所述固相树脂上裂解下来,以得到所述多肽产物。
10.进一步的,所述多肽为具有10-100个氨基酸的多肽,优选为具有20-100个氨基酸的多肽,更优选为具有40-100个氨基酸的多肽。
11.进一步的,步骤(a)中,所述氨基酸临时保护基团为fmoc。
12.进一步的,步骤(a)中,所述固相树脂为王树脂、rink树脂、hmpa-pega树脂、fmpb am树脂或dhp hm树脂。优选为王树脂和rink树脂。
13.进一步的,步骤(a)中,脱除氨基酸临时保护基团的溶液为碱性溶液。特别地,可为仲胺分子的溶液。更具体的说,该碱性溶液可为哌啶的dmf溶液、4-甲基哌啶的dmf溶液、哌啶的n-甲基吡咯烷酮溶液、4-甲基哌啶的n-甲基吡咯烷酮溶液或它们的组合,该碱性溶液
中包含15体积%至25体积%的哌啶或4-甲基哌啶。
14.进一步的,步骤(b)中,所述酸性溶液和碱性溶液的溶剂为二氯甲烷、n-甲基吡咯烷酮、二甲基甲酰胺、二甲亚砜或水。
15.进一步的,步骤(b)中,所述酸性溶液的溶质为三氟乙酸、三氟乙醇、盐酸中的一种以上;其在酸性溶液中的体积分数为0.1%~20%,优选为1%~15%,更优选为5%~10%。
16.进一步的,步骤(b)中,所述碱性溶液的溶质为n,n-二异丙基乙胺、三乙胺、吡啶、氨水中的一种以上;其在碱性溶液中的体积分数为1%~10%,优选为2%~8%,更优选为4%~6%。
17.进一步的,步骤(b)中,依次使用酸性溶液和碱性溶液对固相树脂进行处理时,其具体方式可为浸泡或连续流冲洗,其中在浸泡的同时可以进行振荡、搅拌、鼓泡以及它们的组合。
18.进一步的,步骤(c)中,氨基酸的偶合中使用的缩合试剂选自1-羟基苯并三唑一水合物(hobt)、n,n-二环己基碳二亚胺(dcc)、n,n-二异丙基碳二亚胺(dic)、o-苯并三氮唑-n,n,n',n'-四甲基脲四氟硼酸酯(tbtu)、2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hbtu)、3-(二乙氧基磷酰氧基)-1,2,3-苯并三嗪-4-酮(depbt)中的一种或多种。
19.进一步的,步骤(d)中,裂解液为三氟乙酸、三乙基硅烷、苯甲硫醚和苯酚的混合物,体积比例为85∶5∶5∶5。
20.更近一步的,所述多肽为艾塞那肽类似物,其包括如下步骤:(1)称取键合量为0.5mmol/g 的固相树脂2g,即总键合量为1mmol;向其中加入仲胺分子的溶液,搅拌反应20分钟,脱除fmoc基团;脱除fmoc反应结束后,使用dmf与ipa交替清洗固相树脂;此时向固相树脂中加入2mmol的具有保护基团的氨基酸fmoc-lys(dde)-oh、2mmol tbtu以及4mmol dipea,室温条件下搅拌反应2小时,以偶合氨基酸lys40;偶合反应结束后,使用dmf与ipa交替清洗固相树脂;按照艾塞那肽类似物的肽序列重复上述fmoc脱除和氨基酸连接过程,合成至gly29结束;(2)从合成asn28开始,先加入仲胺分子的溶液,搅拌反应20分钟,用于脱除fmoc基团;脱除fmoc反应结束后,使用dmf与ipa交替清洗固相树脂,清洗完毕后使用酸性溶液清洗固相树脂,然后使用碱性溶液清洗固相树脂,最后使用dmf清洗固相树脂;然后连接氨基酸asn28,此时向固相树脂中加入2 mmol的具有保护基团的氨基酸fmoc-asn(trt)-oh、2 mmol tbtu以及4 mmol dipea,室温条件下搅拌反应2小时;连接反应结束后,使用dmf与ipa交替清洗固相树脂;按照艾塞那肽类似物的肽序列重复上述fmoc脱除和氨基酸连接过程,合成至leu10结束;(3)从asp9开始,重复步骤(1)中的fmoc脱除过程与氨基酸连接过程,按照艾塞那肽类似物的肽序列将不同的氨基酸逐一连接到固相树脂上,主链合成至his1结束;而后采用2%水合肼/dmf处理固相树脂两次各10分钟以脱除dde保护基团,随后的aeeac和mpa的偶合条件为:2 mmol aeeac/mpa、2 mmol hobt以及2 mmol dic,室温下搅拌反应2小时;(4)将步骤(3)中制得的固相树脂干燥后加入到裂解液中,将多肽从固相树脂上裂解下来,同时去除侧链保护基团,再通过析晶干燥得到艾塞那肽类似物粗肽。
21.本发明的有益效果在于:本发明的方法通过依次使用酸性溶液和碱性溶液对固相树脂进行处理,有效提高了多肽产品的纯度,从而降低了后续纯化的困难、提高了多肽固相合成的整体效率。
22.特别是对于一些序列较长、易形成二级结构的多肽序列,本发明的方法能够显著提高固相合成该类多肽产物的纯度。
23.采用本方法制备的多肽产品中潜在杂质的数量和含量都相应减少,从而增强了样品的生物学活性,而对药用多肽来说其安全和有效性都会得到提高。
24.本发明的方法简单易行,在多肽的实验室规模合成和工业化生产中都具有很高的实用价值。
附图说明
25.图1为多肽固相合成技术的基本原理图。
26.图2为根据对比例的常规多肽固相合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
27.图3为根据本发明实施例1的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
28.图4为根据本发明实施例2的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
29.图5为根据本发明实施例3的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
30.图6为根据本发明实施例4的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
31.图7为根据本发明实施例5的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
32.图8为根据本发明实施例6的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
33.图9为根据本发明实施例7的合成方法制备的艾塞那肽类似物粗肽的hplc分析图谱。
具体实施方式
34.以下通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。
35.本发明提供了一种多肽固相合成方法,主要包括以下步骤:1)脱除固相树脂上的氨基酸临时保护基团;2)依次使用酸性溶液和碱性溶液对所述固相树脂进行处理;3)通过逐一缩合的方式按照目标序列依次进行氨基酸的偶合,以合成具有目标序列的多肽;4)将所述具有目标序列的多肽从所述固相树脂上裂解下来,以得到所述多肽产物。
36.本发明所述的“多肽产物”或“粗肽”是指通过多肽的固相合成法直接得到的产物,而未经进一步纯化。该多肽产物可经过诸如高效液相色谱之类的纯化技术进一步纯化,从而得到多肽产品。
37.本发明的多肽产物可为具有10~100个氨基酸的多肽,优选为具有20~100个氨基酸的多肽,更优选为具有40~100个氨基酸的多肽。
38.本发明的方法的步骤1)中使用的固相树脂连接有包含临时保护基团的氨基酸。在本发明的一个具体实施方案中,氨基酸临时保护基团为fmoc。
39.本发明的方法的步骤2)中使用的酸性溶液包含酸和溶剂,其中酸性溶液包含0.1体积%至20体积%的酸,优选包含1体积%至15体积%的酸,更优选包含5体积%至10体积%的酸。当酸含量低于0.1体积%时,酸性溶液由于酸性过低而难以在固相树脂的处理中发挥作用;当酸含量高于20体积%时,酸性过强可能会面临将多肽链从固相树脂上裂解下来的风险。酸可选自三氟乙酸、三氟乙醇、盐酸以及它们的组合,溶剂可选自二氯甲烷、n-甲基吡咯烷酮、二甲基甲酰胺、二甲亚砜和水。
40.本发明的方法的步骤2)中使用的碱性溶液包含碱和溶剂,其中碱性溶液包含1体积%至10体积%的碱,优选包含2体积%至8体积%的碱,更优选包含4体积%至6体积%的碱。当碱含量低于1体积%时,碱性溶液由于碱性过低而难以在固相树脂的处理中发挥作用;当碱含量高于10体积%时,过强的碱性可能会引发一些消旋、天冬酰亚胺副反应等问题。碱可选自n,n-二异丙基乙胺、三乙胺、吡啶、氨水以及它们的组合,溶剂可选自二氯甲烷、n-甲基吡咯烷酮、二甲基甲酰胺、二甲亚砜和水。
41.据目前实验室观察到的数据及相关研究,我们推测使用酸/碱处理能够提高产物纯度的原因,可能是酸碱处理能去除某些在fmoc脱除过程中产生的一些中间产物,使其最大程度上转化为预期的自由氨基,即提高了fmoc脱除的脱除效率,从而减少了相关杂质肽的产生。
42.在本发明的方法的步骤4)中,将具有目标序列的多肽树脂加入到裂解液(tfa、三乙基硅烷、苯甲硫醚和苯酚的混合物,体积比例为85∶5∶5∶5)中反应3h,以得到多肽产物。
43.以下通过一个具体的实施方案进一步解释或说明本发明的内容,但这些例子不应被理解为对本发明的保护范围的限制。
44.在该具体实施方案中,多肽产物可为艾塞那肽类似物[lys40(ε-aeeac-mpa)-nh2]-exendin-4(以下简称cjc-1134,其多肽序列为:h-his-gly-glu-gly-thr-phe-thr-ser-asp-leu-ser-lys-gln-met-glu-glu-glu-ala-val-arg-leu-phe-ile-glu-trp-leu-lys-asn-gly-gly-pro-ser-ser-gly-ala-pro-pro-pro-ser-lys(aeeac-mpa)-nh2)。
[0045]
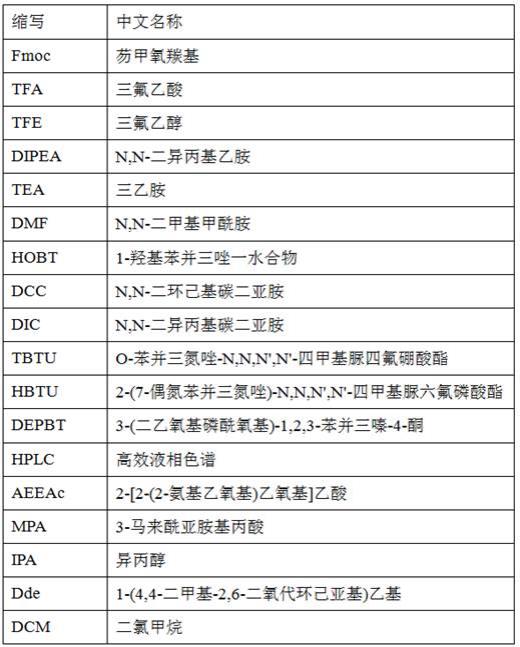
本发明所使用的缩写的含义列于下表1中。
[0046]
表1 缩写名称对照表
。
[0047]
以下对比例/实施例所用材料和仪器的来源如下:固相树脂:氨基树脂(ramage树脂、novasyn tga树脂)和所用保护氨基酸购自bachem;tfa购自阿拉丁;三乙基硅烷、苯甲硫醚、苯酚购自国药集团;tbtu、dipea、hob和dic均购自苏州昊帆;dmf和ipa购自西陇科学;dcm购自山东金岭。
[0048]
高效液相色谱:thermo u3000,分析软件:chromeleon,色谱柱:kromasil c8,流速:1.0 ml/min,柱温:35
˚
c,检测器波长:220 nm,进样体积:20 μl。
[0049]
hplc梯度如下:
。
[0050]
对比例采用常规多肽固相合成方法制备cjc-1134粗肽常规多肽固相合成方法的具体步骤如下:(1)称取键合量为 0.5 mmol/g的固相树脂(氨基树脂,ramage resin)2克,即总键合量为1 mmol。向其中加入哌啶浓度为20体积%的dmf溶液,搅拌反应20分钟,从而脱除fmoc基团。脱除fmoc反应结束后,使用dmf与ipa交替清洗固相树脂。此时向固相树脂中加入2 mmol fmoc-lys(dde)-oh、2 mmol tbtu以及4 mmol dipea,室温条件下搅拌反应2小时,以偶合氨基酸lys40。偶合反应结束后,使用dmf与ipa交替清洗固相树脂。按照cjc-1134的肽序列重复上述fmoc脱除和氨基酸连接过程,主链合成至his1结束。而后采用2%水合肼/dmf处理固相树脂两次各10分钟以去除dde保护基团,随后的aeeac和mpa的偶合条件为,2 mmol aeeac/mpa、2 mmol hobt以及2 mmol dic,室温下搅拌反应2小时。
[0051]
(2)将步骤(1)中的固相树脂干燥后加入到裂解液(tfa、三乙基硅烷、苯甲硫醚和苯酚的混合物,体积比例为85∶5∶5∶5)中,将多肽从固相树脂上裂解下来,同时去除侧链保护基团,再通过析晶干燥得到cjc-1134粗肽。
[0052]
采用常规多肽固相合成方法制备的cjc-1134粗肽的hplc分析图谱如图2所示,hplc曲线的积分结果示于下表2中。由表2中的积分结果可知,对比例中制备的cjc-1134粗肽包含杂质(前杂和后杂)和cjc-1134,其中cjc-1134的峰面积占总峰面积的26.55%,也就是说,常规方法制备的cjc-1134粗肽的纯度为26.55%。
[0053]
表2 对比例中制备的cjc-1134粗肽的hplc曲线积分结果。
[0054]
实施例1
ꢀꢀ
本发明的方法制备cjc-1134粗肽
根据本发明的提高固相合成多肽产物纯度的方法制备cjc-1134粗肽的步骤如下:(1)称取键合量为0.5mmol/g的固相树脂(氨基树脂,ramage树脂)2克,即总键合量为1mmol。向其中加入哌啶浓度为20体积%的dmf溶液,搅拌反应20分钟,从而脱除fmoc基团。脱除fmoc反应结束后,使用dmf与ipa交替清洗固相树脂。此时向固相树脂中加入2mmol的具有保护基团的氨基酸fmoc-lys(dde)-oh、2mmoltbtu以及4mmoldipea,室温条件下搅拌反应2小时,以偶合氨基酸lys40。偶合反应结束后,使用dmf与ipa交替清洗固相树脂。按照cjc-1134的肽序列重复上述fmoc脱除和氨基酸连接过程,合成至gly29结束。
[0055]
(2)从合成asn28开始,先加入哌啶浓度为20体积%的dmf溶液,搅拌反应20分钟,用于脱除fmoc基团。脱除fmoc反应结束后,使用dmf与ipa交替清洗固相树脂,清洗完毕后使用0.1体积%tfa的dcm溶液清洗固相树脂,然后使用5体积%dipea的dcm溶液清洗固相树脂两遍,最后使用dmf清洗固相树脂。然后连接氨基酸asn28,此时向固相树脂中加入2mmol的具有保护基团的氨基酸fmoc-asn(trt)-oh、2mmoltbtu以及4mmoldipea,室温条件下搅拌反应2小时。连接反应结束后,使用dmf与ipa交替清洗固相树脂。按cjc-1134的肽序列重复上述fmoc脱除和氨基酸连接过程,合成至leu10结束。
[0056]
(3)从asp9开始,重复步骤(1)中的fmoc脱除过程与氨基酸连接过程,按照cjc-1134的肽序列将不同的氨基酸逐一连接到固相树脂上,主链合成至his1结束。尔后采用2%水合肼/dmf处理固相树脂两次各10分钟以脱除dde保护基团,随后的aeeac和mpa的偶合条件为:2mmolaeeac/mpa、2mmolhobt以及2mmoldic,室温下搅拌反应2小时。
[0057]
(4)将步骤(3)中的固相树脂干燥后加入到裂解液(tfa、三乙基硅烷、苯甲硫醚和苯酚的混合物,体积比例为85∶5∶5∶5)中,将多肽从固相树脂上裂解下来,同时去除侧链保护基团,再通过析晶干燥得到cjc-1134粗肽。
[0058]
实施例1中制备的cjc-1134粗肽的hplc分析图谱如图3所示,hplc曲线的积分结果示于下表3中。
[0059]
表3实施例1中制备的cjc-1134粗肽的hplc曲线积分结果。
[0060]
由表3中的积分结果可知,实施例1中制备的cjc-1134粗肽包含杂质(前杂和后杂)和cjc-1134,其中cjc-1134的峰面积占总峰面积的34.42%,从而表明实施例1的方法制备的cjc-1134粗肽的纯度为34.42%。与对比例的常规方法相比,实施例1的方法使cjc-1134粗肽的纯度提高了30%。
[0061]
实施例2本发明的方法制备cjc-1134粗肽实施例2的方法步骤与实施例1类似,不同之处在于,在步骤(1)和步骤(2)中使用4-甲基哌啶浓度为20体积%的dmf溶液脱除fmoc基团,以及在步骤(2)中使用10体积%tfe的
dcm溶液清洗固相树脂,然后使用5体积%tea的dcm溶液清洗固相树脂两遍。
[0062]
实施例2中制备的cjc-1134粗肽的hplc分析图谱如图4所示,hplc曲线的积分结果示于下表4中。
[0063]
表4实施例2中制备的cjc-1134粗肽的hplc曲线积分结果。
[0064]
由表4中的积分结果可知,实施例2中制备的cjc-1134粗肽包含杂质(前杂和后杂)和cjc-1134,其中cjc-1134的峰面积占总峰面积的34.12%,从而表明实施例2的方法制备的cjc-1134粗肽的纯度为34.12%。与对比例的常规方法相比,实施例2的方法使cjc-1134粗肽的纯度提高了29%。
[0065]
实施例3本发明的方法制备cjc-1134粗肽实施例3的方法步骤与实施例1类似,不同之处在于,在步骤(2)中使用1体积%tfa的dcm溶液清洗固相树脂,然后使用10体积%dipea的dcm溶液清洗固相树脂两遍。
[0066]
实施例3中制备的cjc-1134粗肽的hplc分析图谱如图5所示,hplc曲线的积分结果示于下表5中。
[0067]
表5实施例3中制备的cjc-1134粗肽的hplc曲线积分结果。
[0068]
由表5中的积分结果可知,实施例3的方法制备的cjc-1134粗肽的纯度为32.80%。与对比例的常规方法相比,实施例3的方法使cjc-1134粗肽的纯度提高了24%。
[0069]
实施例4本发明的方法制备cjc-1134粗肽实施例4的方法步骤与实施例1类似,不同之处在于,在步骤(2)中使用5体积%tfe的dcm溶液清洗固相树脂,然后使用2体积%tea的dcm溶液清洗固相树脂两遍。
[0070]
实施例4中制备的cjc-1134粗肽的hplc分析图谱如图6所示,hplc曲线的积分结果示于下表6中。
[0071]
表6实施例4中制备的cjc-1134粗肽的hplc曲线积分结果
。
[0072]
由表6中的积分结果可知,实施例4的方法制备的cjc-1134粗肽的纯度为34.86%。与对比例的常规方法相比,实施例4的方法使cjc-1134粗肽的纯度提高了31%。
[0073]
实施例5本发明的方法制备cjc-1134粗肽实施例5的方法步骤与实施例1类似,不同之处在于,在步骤(2)中使用20体积%tfe的dcm溶液清洗固相树脂,然后使用10体积%tea的dcm溶液清洗固相树脂两遍。
[0074]
实施例5中制备的cjc-1134粗肽的hplc分析图谱如图7所示,hplc曲线的积分结果示于下表7中。
[0075]
表7实施例5中制备的cjc-1134粗肽的hplc曲线积分结果。
[0076]
由表7中的积分结果可知,实施例5的方法制备的cjc-1134粗肽的纯度为34.93%。与对比例的常规方法相比,实施例5的方法使cjc-1134粗肽的纯度提高了32%。
[0077]
实施例6本发明的方法制备cjc-1134粗肽实施例6的方法步骤与实施例1类似,不同之处在于,在步骤(2)中使用25体积%tfe的dcm溶液清洗固相树脂,然后使用15体积%tea的dcm溶液清洗固相树脂两遍。
[0078]
实施例6中制备的cjc-1134粗肽的hplc分析图谱如图8所示,hplc曲线的积分结果示于下表8中。
[0079]
表8实施例6中制备的cjc-1134粗肽的hplc曲线积分结果
。
[0080]
由表8中的积分结果可知,实施例6的方法制备的cjc-1134粗肽的纯度为27.96%。与对比例的常规方法相比,实施例6的方法使cjc-1134粗肽的纯度提高了5%。
[0081]
实施例7本发明的方法制备cjc-1134粗肽实施例7的方法步骤与实施例1类似,不同之处在于,在步骤(1)中使用的固相树脂为novasyntga树脂,在步骤(2)中使用0.1体积%盐酸的水溶液清洗固相树脂,然后使用0.2体积%氨水的水溶液清洗固相树脂两遍。
[0082]
实施例7中制备的cjc-1134粗肽的hplc分析图谱如图9所示,hplc曲线的积分结果示于下表9中。
[0083]
表9实施例7中制备的cjc-1134粗肽的hplc曲线积分结果。
[0084]
由表9中的积分结果可知,实施例7的方法制备的cjc-1134粗肽的纯度为27.96%。与对比例的常规方法相比,实施例7的方法使cjc-1134粗肽的纯度提高了5%。
[0085]
综合上述各实施例,下表10示出了各实施例的步骤(2)中使用的酸性溶液和碱性溶液的溶质和溶剂。
[0086]
表10各实施例的步骤(2)中使用的酸性溶液和碱性溶液
。
[0087]
实施例1~7中制备的cjc-1134粗肽的hplc曲线分析结果总结于下表11中,从hplc曲线的积分结果可以看到,与实施例1和2类似地,实施例3~5中制备的cjc-1134粗肽的纯度均为约34%,表明本发明的方法将多肽产物(粗肽)纯度由常规方法的26.55%大幅提高到了34%左右,提高幅度高达约30%,具有非常显著的提高多肽产物纯度的效果。在实施例6中,可能是酸性溶液和碱性溶液的浓度过大导致了其他副反应的产生,多肽产物的纯度与常规方法相比仅有小幅提高。在实施例7中,虽然粗肽纯度提高的幅度没有实施例1~5中高,但也达到了15%。
[0088]
表11各实施例中制备的cjc-1134粗肽的hplc曲线的积分结果。
[0089]
通过本发明的方法,使固相合成多肽产物的纯度得到了显著提高,这种程度的纯度提高将为后续的多肽纯化步骤带来极大的便利,也为多肽整体生产效率的提高奠定了基础。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1