一种水溶性抗肿瘤前药及其药物组合物和用途的制作方法
本发明是申请日为2021年8月23日、申请号为202110970500.8、发明名称为“一种水溶性抗肿瘤前药及其药物组合物和用途”专利的分案申请。
背景技术:
1、癌症是威胁人类健康的第二大疾病,从全球情况看,近六分之一的死亡由癌症造成,癌症负担不断加重。
2、癌症的治疗包括手术治疗、放射治疗、化疗、新兴的分子靶向治疗和免疫治疗等。在精准医学的时代背景下,开发疗效高、广谱性强、副作用小的抗肿瘤药物是新药研发的重要方向。其中,以细胞毒药物为基础的化疗因其疗效显著、广谱性强等特点在恶性肿瘤尤其是实体瘤的综合治疗中具有重要地位。如作为术前和术后的辅助化疗或新辅助化疗对于预防癌症的复发和转移具有不可替代的作用;此外,通过对晚期癌症患者进行姑息性化疗可以控制癌症的发展以延长生命,改善生存质量;另一方面,化疗药物联合分子靶向药物和免疫治疗药物治疗是临床恶性肿瘤或耐药肿瘤治疗的常用手段。
3、但是传统的化疗药物水溶性差、对肿瘤和正常组织缺乏选择性,仍然面临着诸如制剂工艺复杂、治疗窗口狭窄、耐药性增加和非特异性毒性等限制因素,从而导致临床治疗效果不佳且影响患者的生存质量和接受程度。
4、喜树碱(cpt)为一种吡咯喹啉细胞毒性生物碱,是除紫杉醇之外,研究最多的天然抗肿瘤药物之一。研究表明其对胃癌、直肠癌和白血病等多种恶性肿瘤均有一定的疗效。但是由于该生物碱易产生恶心、呕吐、腹泻、脱发等副作用,且其水溶性差,制成水溶性的钠盐后抗肿瘤活性降低。喜树碱及其衍生物是以拓朴异构酶(topo i)为作用靶点抑制dna的合成而发挥抗癌作用的机理,研发高效低毒的喜树碱衍生物,成为抗癌领域研究的新热点。
5、伊立替康(irinotecan,cpt-11)是喜树碱类中上市的新药物之一,由日本第一制药公司研制开发,是治疗转移性结直肠癌的有效药物,对氟尿嘧啶耐药病例仍有效。伊立替康的常见副作用包括:迟发性腹泻(用药24小时后发生),中性粒细胞减少,恶心与呕吐,急性胆碱能综合征(包括早发性腹泻,用药24小时内发生);少数轻微反应有:厌食、腹痛、黏膜炎、乏力、血小板减少症。其中约有20%以上的患者在接受以伊立替康为基础的联合化疗方案时出现严重的中性粒细胞减少和/或腹泻。伊立替康的毒性主要是由其活性代谢产物sn-38(7-乙基-10-羟基喜树碱)引起的。sn-38主要与血浆蛋白结合(结合率95%),在发挥完抗肿瘤作用后,活性sn-38主要是通过位于肝脏的ugt1a1(尿苷二磷酸葡萄糖醛酸基转移酶iai)的催化作用而转变为无活性的sn-38g(活性约为sn-38的1/50~1/100),后者再通过尿液、胆汁排出;同时ugt1a1亦可参与胆红素的糖基化转换,进而产生溶解性更高的结合胆红素。
6、5氟尿嘧啶(5-fu)是一个抗代谢且在临床上是应用最广的嘧啶类抗肿瘤药物,对消化道癌及其他实体瘤有良好疗效,在肿瘤内科治疗中占有重要地位。它需经过代谢为5-氟脱氧尿嘧啶核苷酸而具有抗肿瘤活性。5-fu通过抑制胸腺嘧啶核苷酸合成酶而抑制dna的合成。主要的副作用有骨髓抑制,为白细胞减少、血小板下降;食欲不振、恶心、呕吐、口腔炎、胃炎、腹痛及腹泻等胃肠道反应;注射局部有疼痛、静脉炎或动脉内膜炎;常有脱发、红斑性皮炎、皮肤色素沉着手足综合征及暂时性小脑运动失调,偶有影响心脏功能。
7、氮芥类药物是β-氯乙胺类化合物的总称,其结构可分为两部分:烷基化部分和载体部分。烷基化部分(即双β-氯乙胺基,也称氮芥基)是抗肿瘤活性的功能基团;载体部分主要影响药物在体内的吸收、分布等药代动力学性质,通过选择不同的载体,可以达到提高药物选择性和疗效、降低毒性的目的。氮芥类化合物由于毒性太大而不能直接药用,可对其结构进行改造得到氮芥类抗肿瘤药物。
8、因此,为了克服上述障碍,本领域迫切需要开发合成一种既能改善化合物水溶性,也能降低对正常细胞毒性,提高抗肿瘤效果更好的化疗药物。
技术实现思路
0、发明概述
1、以下是对本文详细描述的主题的概述。本概述并非是为了限制本技术的保护范围。
2、本技术提供了一种水溶性抗肿瘤药物的前药,所述的水溶性抗肿瘤药物的前药如式(i)所示,或其互变异构体、或立体异构体、或药学上可接受的盐:
3、
4、式(i)中m为:
5、
6、这里,r1为:
7、
8、
9、l为-c(=o)-、-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即以m和r1中一方的基团相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;
10、或者,l-r1为-p(=o)(r1)2、-p(=o)(r1)(oh)、或
11、
12、其中,式(i-a)中r2选自氢、n,n-二甲胺基甲基、硝基和c1-c6烷基;r3选自氢和c1-c6烷基;
13、式(i-b)中r4选自氢、或如下结构:
14、式(i-i)中r5和r6各自独立地选自氢和羟基,或者,r5和r6与其相连的碳原子一起组成如下结构:这里,r19和r20各自独立选自氢、c1-c4烷基、苯基、取代的苯基,所述取代的苯基是指苯环上的氢任选地被一个或多个甲氧基、硝基和卤素所取代;r7为氢、甲基或羟甲基;
15、式(i-c)中r8为不存在(即直接与n原子相连)、或-ph-(ch2)n-c(=o)-;这里,ph为苯,n为1-4的整数;
16、式(i-d)中r9、r10、r11和r12各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;x-为有机酸或无机酸的酸根;
17、式(i-e)中r13、r14、r15、r16和r17各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;r13为氢、或c1-c6烷基;
18、式(i-f)、式(i-g)和式(i-h)中y4和y5各自独立地为不存在(或键)、或c1-c6亚烷基;r18为氢、羟基、c1-c6烷氧基、或c1-c6烷基;r1的定义如上;
19、并规定,式(i)化合物不包括下列化合物:
20、及其盐。
21、另一方面,本技术提供了上述水溶性抗肿瘤药物的前药的制备方法,所述的制备方法包括如下步骤:
22、式m-lwg1化合物与式lwg2-l-r1化合物发生偶联反应,得到式(i)化合物;
23、这里,式m-lwg1化合物、式lwg2-l-r1化合物和式(i)化合物中m、l以及r1的定义如上;lwg1和lwg2各自独立地为离去基团。
24、第三方面,本技术提供了包含上述水溶性抗肿瘤药物的前药的药物组合物。
25、第四方面,本技术提供了上述水溶性抗肿瘤药物的前药及其药物组合物抗肿瘤的用途。
26、在本技术的一些实施方式中,本技术提供了一种如式(i)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
27、所述的式(i)化合物为式(i-1)化合物:
28、
29、这里,r1为:
30、
31、l为-c(=o)-、-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即以m和r1中一方的基团相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;
32、或者,l-r1为-p(=o)(r1)2、-p(=o)(r1)(oh)、或
33、
34、其中,式(i-1)中r2选自氢、n,n-二甲胺基甲基、硝基和c1-c6烷基;r3选自氢和c1-c6烷基;
35、式(i-d)中r9、r10、r11和r12各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;x-为有机酸或无机酸的酸根;
36、式(i-e)中r13、r14、r15、r16和r17各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;r13为氢、或c1-c6烷基;
37、式(i-f)、式(i-g)和式(i-h)中y4和y5各自独立地为不存在(或键)、或c1-c6亚烷基;r18为氢、羟基、c1-c6烷氧基、或c1-c6烷基;r1的定义如上。
38、在本技术的一些实施方式中,本技术提供了一种如式(i-1)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
39、其中,式(i-1)中r2选自氢、n,n-二甲胺基甲基、硝基和乙基;r3选自氢和乙基。
40、在本技术的一些实施方式中,本技术提供了一种如式(i-1)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
41、其中,式(i-1)中r2为氢;r3为乙基。
42、在本技术的一些实施方式中,本技术提供了一种如式(i-1)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
43、其中,式(i-1)中r2为n,n-二甲胺基甲基;r3为氢。
44、在本技术的一些实施方式中,本技术提供了一种如式(i-1)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
45、其中,式(i-1)中r2为硝基;r3为氢。
46、在本技术的一些实施方式中,本技术提供了一种如式(i-1)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
47、其中,式(i-1)中r2为氢;r3为氢。
48、在本技术的一些实施方式中,本技术提供了一种如式(i)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;所述的式(i)化合物为式(i-2)化合物:
49、
50、这里,r1为:
51、
52、l为-c(=o)-、-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即以m和r1中一方的基团相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;
53、或者,l-r1为-p(=o)(r1)2、-p(=o)(r1)(oh)、或
54、
55、式(i-2)中r4选自氢、或如下结构:
56、式(i-i)中r5和r6各自独立地选自氢和羟基,或者,r5和r6与其相连的碳原子一起组成如下结构:这里,r19和r20各自独立选自氢、c1-c4烷基、苯基、取代的苯基,所述取代的苯基是指苯环上的氢任选地被一个或多个甲氧基、硝基和卤素所取代;r7为氢、甲基或羟甲基;
57、式(i-d)中r9、r10、r11和r12各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;x-为有机酸或无机酸的酸根;
58、式(i-e)中r13、r14、r15、r16和r17各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;r13为氢、或c1-c6烷基;
59、式(i-f)、式(i-g)和式(i-h)中y4和y5各自独立地为不存在(或键)、或c1-c6亚烷基;r18为氢、羟基、c1-c6烷氧基、或c1-c6烷基;r1的定义如上。
60、在本技术的一些实施方式中,本技术提供了一种如式(i-2)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
61、其中,式(i-i)中r5和r6各自独立地为羟基;r7为甲基。
62、在本技术的一些实施方式中,本技术提供了一种如式(i-2)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
63、其中,式(i-i)中r5和r6各自独立地为氢;r7为氢。
64、在本技术的一些实施方式中,本技术提供了一种如式(i-2)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
65、其中,式(i-i)中r5为氢;r6为羟基;r7为羟甲基。
66、在本技术的一些实施方式中,本技术提供了一种如式(i-2)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
67、其中,r5和r6与其相连的碳原子一起组成如下结构:这里,r19和r20都是甲基;r7为甲基或羟甲基。
68、在本技术的一些实施方式中,本技术提供了一种如式(i-2)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
69、其中,r5和r6与其相连的碳原子一起组成如下结构:这里,r19是氢而r20是苯基;r7为甲基或羟甲基。
70、在本技术的一些实施方式中,本技术提供了一种如式(i)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;所述的式(i)化合物为式(i-3)化合物:
71、
72、这里,r1为:
73、
74、l为-c(=o)-、-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即以m和r1中一方的基团相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;
75、或者,l-r1为-p(=o)(r1)2、-p(=o)(r1)(oh)、或
76、
77、式(i-3)中r8为不存在(即直接与n原子相连)、或-ph-(ch2)n-c(=o)-;这里,ph为苯,n为1-4的整数;
78、式(i-d)中r9、r10、r11和r12各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;x-为有机酸或无机酸的酸根;
79、式(i-e)中r13、r14、r15、r16和r17各自独立地为氢、氘、c1-c6烷基、c1-c6烷氧基、c1-c8烷酰基、c3-c7环烷基、羟基、氨基、胺基、或氨基酸;r13为氢、或c1-c6烷基;
80、式(i-f)、式(i-g)和式(i-h)中y4和y5各自独立地为不存在(或键)、或c1-c6亚烷基;r18为氢、羟基、c1-c6烷氧基、或c1-c6烷基;r1的定义如上;
81、并规定,式(i-3)化合物不包括下列化合物:
82、及其盐。
83、在本技术的一些实施方式中,本技术提供了一种如式(i-3)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
84、其中,式(i-3)中r8为不存在(即直接与n原子相连);
85、并规定,式(i-3)化合物不包括下列化合物:
86、及其盐。
87、在本技术的一些实施方式中,本技术提供了一种如式(i-3)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
88、其中,式(i-3)中r8为-ph-(ch2)n-c(=o)-;这里,-ph-为1,4-亚苯基,n为1-4的整数。
89、在本技术的一些实施方式中,本技术提供了一种如式(i-3)所示水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐;
90、其中,式(i-3)中r8为-ph-(ch2)n-c(=o)-;这里,-ph-为1,4-亚苯基,n为2或3。
91、在本技术的实施方式中,所述的氨基酸可选自天然氨基酸即l-氨基酸,或非天然氨基酸,所述的氨基酸为甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、甲硫氨酸、脯氨酸、色氨酸、丝氨酸、酪氨酸、半胱氨酸、苯丙氨酸、天冬酰胺、谷氨酰胺、苏氨酸、天冬氨酸、谷氨酸、赖氨酸、精氨酸或组氨酸。
92、在本技术的实施方式中,所述的c1-c6烷基可选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基和己基等。
93、在本技术的实施方式中,所述的c1-c7酰基可选自甲酰基、乙酰基、丙酰基、正丁酰基和异丁酰基等。
94、在本技术的实施方式中,所述的c3-c7环烷基可选自环丙烷基、环丁烷基、环戊烷基、环己烷基和环庚基。
95、在本技术的实施方式中,所述的卤原子选自f、cl、br、i。
96、在本技术的实施方式中,-c(=o)-y1-c(=o)-可以是乙二酰基、丙二酰基、丁二酰基、丁烯二酰基、羟基丁二酰基、氨基丁二酰基、戊二酰基、戊烯二酰基,羟基戊二酰基、氨基戊二酰基、己二酰基、己烯二酰基,羟基己二酰基、氨基己二酰基、庚二酰基、庚烯二酰基,羟基庚二酰基、氨基庚二酰基、辛二酰基、辛烯二酰基,羟基辛二酰基、或氨基辛二酰基。
97、在本技术的一些实施方式中,所述的式(i)化合物为下列结构的化合物,或其互变异构体、或立体异构体、或药学上可接受的盐:
98、
99、
100、式(4)、式(5)、式(6)、式(6’)、式(7)、式(7’)、式(8)、式(9)和式(11)中:l为-c(=o)-、-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即l相邻的两个基团直接相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;
101、式(4)、式(6)、式(6’)和式(8)中:x-为有机酸或无机酸的酸根;
102、式(10)中:l为-c(=o)-y1-c(=o)-、-c(oy2)(oy3)-、-c(oy2)(oy3)-y1-c(oy2)(oy3)-、-c(oy2)h-、-c(oy2)h-y1-c(oy2)(oy3)-、-c(oy2)h-y1-c(oy2)h-、-c(oy2)(oy3)-y1-c(oy2)h-、或-s(=o)2-、或l不存在(即以m和r1中一方的基团相连);其中,y1为不存在、未取代的c1-c6亚烷基、被a组基团取代的c1-c6亚烷基、未取代的c2-c6亚烯基、或被a组基团取代的c2-c6亚烯基;y2和y3各自独立地选自未取代的c1-c6烷基和被a组基团取代的c1-c6烷基;所述的a组基团选自氨基和羟基;x-为有机酸或无机酸的酸根,优选地,为氯离子;
103、或者,l为磷酸酯键、或柠檬酸酯键,任选地,可以连接1个或2个下列基团:
104、这里,x-为有机酸或无机酸的酸根。
105、在本技术的一些实施方式中,式(4)、式(5)、式(6)、式(6’)、式(7)、式(7’)、式(8)、式(9)和式(11)中l为-c(=o)-、乙二酰基、丙二酰基、丁二酰基、丁烯二酰基、羟基丁二酰基、氨基丁二酰基、戊二酰基、戊烯二酰基,羟基戊二酰基、氨基戊二酰基、己二酰基、己烯二酰基,羟基己二酰基、氨基己二酰基、庚二酰基、庚烯二酰基,羟基庚二酰基、氨基庚二酰基、辛二酰基、辛烯二酰基,羟基辛二酰基、或氨基辛二酰基。
106、在本技术的一些实施方式中,式(10)中l为乙二酰基、丙二酰基、丁二酰基、丁烯二酰基、羟基丁二酰基、氨基丁二酰基、戊二酰基、戊烯二酰基,羟基戊二酰基、氨基戊二酰基、己二酰基、己烯二酰基,羟基己二酰基、氨基己二酰基、庚二酰基、庚烯二酰基,羟基庚二酰基、氨基庚二酰基、辛二酰基、辛烯二酰基,羟基辛二酰基、或氨基辛二酰基。
107、在本技术的一些实施例中,所述的式(i)化合物为下列结构的化合物(即化合物a、b、c、e、e’、f、或h):
108、
109、
110、在本技术的实施方式中,本发明的部分化合物分子中含有季铵基团或磺酸基团,可如本专业公认的那样,通过酸处理或碱处理,转化成药学上可接受的盐。本发明所述的药学上可接受的盐包括无机酸盐或有机酸盐,包括与有机和无机的酸或碱生成的盐。这里,所述药学上可接受的酸成盐包括与以下酸形成的盐:盐酸、氢溴酸、硫酸、柠檬酸、酒石酸、磷酸、乳酸、丙酮酸、醋酸、三氟醋酸、丁二酸、乙二酸、富马酸、马来酸、丁酮二酸、甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸和羟乙磺酸。药学上可接受的碱盐包括铵盐、碱金属盐(例如钠盐和钾盐)、碱土金属盐(例如钙盐和镁盐)以及与有机碱(例如二环己基胺和n-甲基-d-葡糖胺)形成的盐。
111、本技术提供了上述水溶性抗肿瘤药物的前药的制备方法,所述的制备方法包括如下步骤:
112、式m-lwg1化合物与式lwg2-l-r1化合物发生偶联反应,得到式(i)化合物;
113、这里,式m-lwg1化合物、式lwg2-l-r1化合物和式(i)化合物中m、l以及r1的定义如上;lwg1和lwg2各自独立地为离去基团。
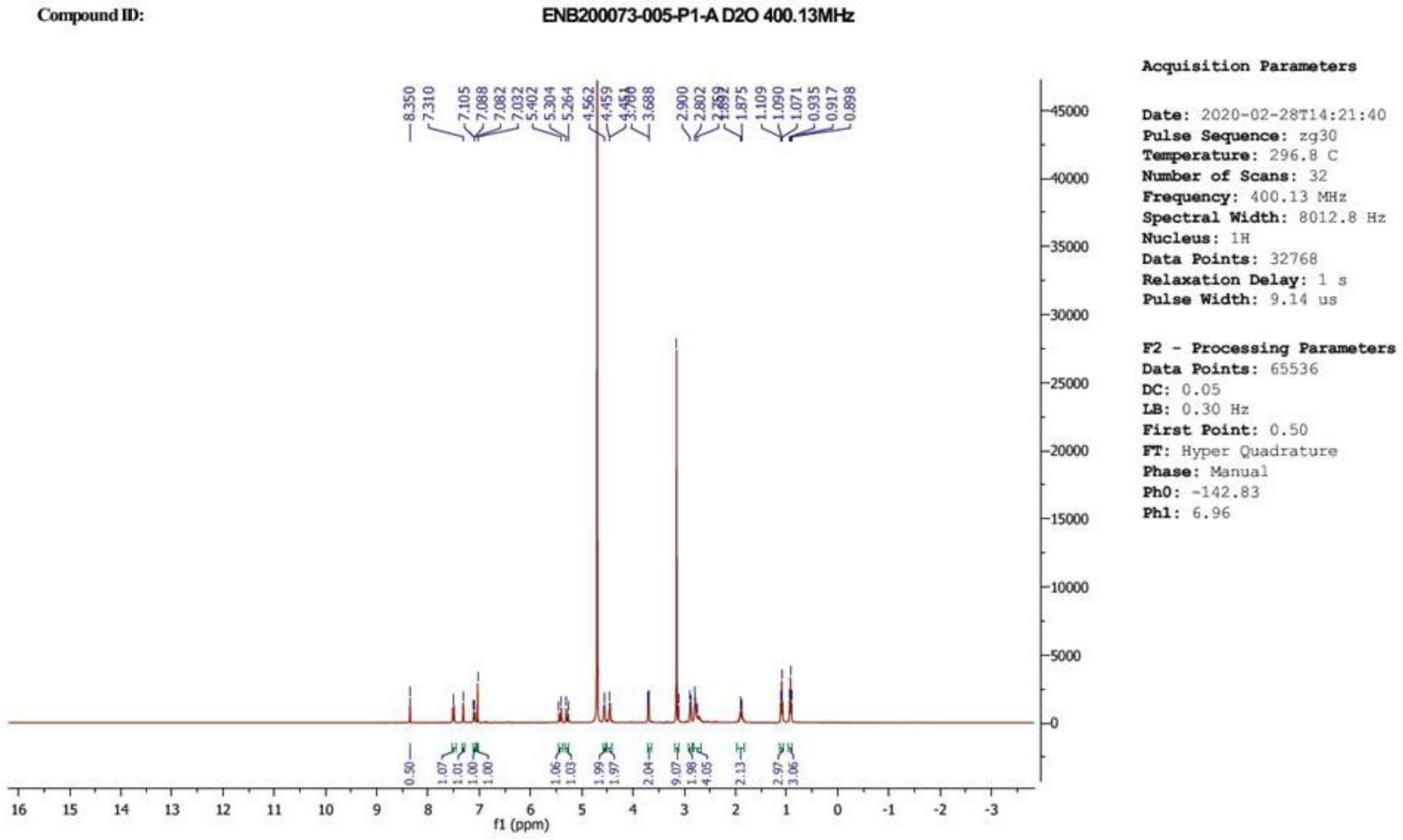
114、在本技术的实施方式中,本技术提供了一种药物组合物,该药物组合物包含治疗有效量的上述水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐,以及药学上可接受的载体。
115、在本技术的一些实施方式中,所述药物组合物含有作为活性成分的化合物a、b、c、e、e’、f、或h,或其互变异构体、或立体异构体、或药学上可接受的盐,以及药学上可接受的载体或赋形剂。
116、所述的药物组合物可根据本领域公知的方法制备不同的非肠道给药剂型,适于人或动物使用。例如,将本发明化合物制成注射用制剂,如溶液剂、混悬剂溶液剂、乳剂、冻干粉针剂,这种制剂可以是含水或非水的,可含一种和多种药效学上可接受的载体、稀释剂、防腐剂、表面活性剂、助溶剂、缓冲剂、ph调节剂。这些辅料都是本领域常用的。为达到用药目的,增强治疗效果,本发明药物或药物组合物可用任何公知的给药方法给药。
117、本发明的组合物可单独服用,或与其他治疗药物或对症药物合并使用,当本发明的化合物与其他治疗药物存在协同作用时,应根据实际情况调整它的剂量。
118、在本技术的实施方式中,本技术提供了水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐,或者其药物组合物抗肿瘤的用途;这里,所述的肿瘤包括肺癌、乳腺癌、卵巢癌、肝癌、结肠癌、和胃癌等。
119、在本技术的实施方式中,本技术提供了水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐,或者其药物组合物治疗肿瘤的方法,包括对肿瘤患者施用治疗有效量的水溶性抗肿瘤药物的前药,或其互变异构体、或立体异构体、或药学上可接受的盐,或者其药物组合物;这里,所述的肿瘤包括肺癌、乳腺癌、卵巢癌、肝癌、结肠癌、和胃癌等。
120、有益技术效果:
121、本发明水溶性抗肿瘤药物的前药不仅提高了水溶性(如化合物b在水中的溶解度是40mg/ml,相比10羟基喜树碱的38μg/ml提高了1000倍)。此外,具有以下独特的优点:分子量小、无免疫原性、廉价易得、稳定性好、与药物分子之间化学键连接简单易行,在体内释放后无毒副作用,应用范围广泛等。
122、该类抗肿瘤前药使得化合物的细胞毒性明显降低,同时,由于肿瘤细胞的快速增殖和高的羧酸酯酶表达,上述药物作为前药被肿瘤细胞摄取并释放活性药物的量远大于正常细胞,从而使活性化合物在肿瘤部位聚集,进而到达抗肿瘤的效果同时减弱对正常器官的毒性,因而可以制成更低毒和有效的抗肿瘤药物。
123、本技术的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本技术而了解。本技术的其他优点可通过在说明书以及附图中所描述的方案来实现和获得。
124、附图概述
125、附图用来提供对本技术技术方案的理解,并且构成说明书的一部分,与本技术的实施例一起用于解释本技术的技术方案,并不构成对本技术技术方案的限制。
126、图1为本技术实施例化合物a的合成1h-nmr谱图;
127、图2为本技术实施例化合物b的合成1h-nmr谱图;
128、图3为本技术实施例化合物e’的合成1h-nmr谱图;
129、图4为本技术实施例化合物h的合成1h-nmr谱图;
130、图5为化合物浓度-细胞活性曲线图;
131、图6为本技术实施例化合物e1s49559(a、d)、sn-38(b、e)和内标yltk(c、f)的质谱及二级质谱图;
132、图7为本技术实施例化合物e1s49559(a)、内标yltk(b)和sn-38(c)以及e1s49559与大鼠血浆孵育后(d)的lc-ms/ms分析的色谱图。
133、图8为本技术实施例化合物h(2020-sxpt-004)标准品溶液的eic图(a);rt(保留时间)8.34min的质谱图(b);rt 8.34min的质谱图局部放大(c);m/z 389.1757的ms/ms谱(d)。
134、图9为本技术实施例化合物h(2020-sxpt-004)的代谢产物1(苯丁酸氮芥)的eic图(a);rt 10.93min的质谱图(b);rt 10.93min的质谱图局部放大(c);m/z 304.0866的ms/ms谱(d)。
135、图10为本技术实施例化合物e’(2020-sxpt-005)的的eic图(a);rt 4.41min的质谱图(b);rt 4.41min的质谱图局部放大(c);m/z 415.1987的ms/ms谱(d)。
136、图11为本技术实施例化合物e’(2020-sxpt-005)的代谢产物m4(5-氟尿嘧啶)的eic图(a);rt 5.35min的质谱图(b);rt 5.35min的质谱图局部放大(c);m/z 130.0411的ms/ms谱(d)。
137、详述
138、下文中将对本发明的实施例进行详细说明。需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互任意组合。
139、缩写:
140、dmf为二甲基甲酰胺;
141、edci为1-乙基-3-(3-二甲基氨基丙基)碳酰二亚胺;
142、dmap为4-二甲氨基吡啶;
143、diea为n,n-二异丙基乙基胺;
144、sn-38为7-乙基-10-羟基喜树碱;
145、mc38为小鼠mc38结肠癌模型;
146、irtw(%)瘤重抑制率;
147、i.p.腹腔注射;
148、bnpp为双-p-硝基苯基磷酸盐;
149、yltk为伊立替康;
150、lc-ms/ms为液相色谱串联质谱分析;
151、lc-ms/hrms为液相色谱串联高分辨质谱分析
152、esi为电喷雾离子化
153、mrm为多反应监测;
154、rt为保留时间;
155、dp为解簇电压;
156、ce为碰撞能量
157、eic为提取离子流图
158、fa为甲酸
159、prm为平行反应监测
160、实施例1:化合物a的合成:
161、化合物1的合成
162、
163、将7-乙基-10-羟基喜树碱(sn-38)(2g,5.1mmol,1.0eq.)溶于四氢呋喃(thf)和二氯甲烷(dcm)的混合溶液(体积比为1∶1,400ml)中,然后依次加入diea(1.7ml,10.2mmol,2.0eq.)和4-硝基苯基氯甲酸酯(2.05g,10.2mmol,2.0eq.)。反应在室温下搅拌24小时。tlc(石油醚/乙酸乙酯(v/v)=1/4)点板,原料反应完全。反应液抽滤后得到化合物1粗品(2g,70.34%)为白色固体。
164、lcms(安捷伦6550质谱仪):[m+h]+=558.3
165、化合物a的合成
166、
167、将化合物1(2g,3.6mmol,1.0eq.)溶于dmf(100ml)中,然后依次加入氯化胆碱(0.75g,7.2mmol,2.0eq.)和diea(0.93g,7.2mmol,2.0eq.)。反应液在氮气保护下,室温搅拌24小时。反应液抽滤并用乙酸乙酯(2x200ml)对滤饼进行重结晶,得到粗产物。粗产物用硅胶快速色谱法(二氯甲烷/甲醇=0~25体积%)纯化后得到产物化合物a。或者将上述粗产物用pre-hplc(0.05%甲酸/乙腈/水)纯化后得到产物化合物a。
168、lcms(安捷伦6550质谱仪):[m]+=522.3
169、实施例2:化合物b的合成:
170、化合物2的合成
171、
172、将丁二酸酐(5g,50mmol,1.0eq.)和二甲氨基乙醇(5.35g,60mmol,1.2eq.)溶于丙酮(100ml)中。反应液在75℃下回流3小时。冷却到室温后,减压浓缩。残留物用丙酮(10ml)重结晶后得到化合物2(8g,产率84.6%)为白色固体。
173、1h nmr(400mhz,cdcl3)δ12.84(br.s,1h),4.31-4.24(m,2h),2.88-2.82(m,2h),2.66-2.57(m,4h),2.46(s,6h).
174、化合物3的合成
175、
176、将化合物2(8g,42.3mmol,1.0eq.)溶于丙酮(100ml)中,然后加入碘甲烷(30g,211.5mmol,5.0eq.)。反应液在75℃下回流24小时。冷却到室温后,抽滤得到化合物3(7g,产率50%)为白色固体。
177、1h nmr(400mhz,d2o)δ4.45-4.42(m,2h),3.61-3.59(m,2h),3.06(s,9h),2.59-2.57(m,4h).
178、化合物b(简称e1s49559)的合成
179、
180、将化合物3(3.38g,10.2mmol,2.0eq.)溶于dmf(40ml)中,然后依次加入edci(2.93g,15.3mmol,3.0eq.),dmap(0.31g,2.5mmol,0.5eq.)和diea(4.2ml,25.5mmol,5.0eq.)。反应液室温搅拌10分钟后,再加入7-乙基-10-羟基喜树碱(2g,5.1mmol,1.0eq.)。反应液在室温下搅拌10小时。反应液用乙酸乙酯(200ml)稀释。反应液抽滤后得到的粗产品用pre-hplc(0.1%甲酸/乙腈/水)纯化,最终得到化合物b(202mg,5.88%)为白色固体。
181、1h nmr(瓦里安,400mhz,d2o)δ7.50(d,j=9.2hz,1h),7.31(d,j=2.0hz,1h),7.10(dd,j=9.2,2.4hz,1h),7.03(s,1h),5.42(d,j=16.0hz,1h),5.28(d,j=16.0hz,1h),4.59-4.54(m,2h),4.46-4.40(m,2h),3.72-3.65(m,2h),3.13(s,9h),2.90-2.88(m,2h),2.80-2.66(m,4h),1.96-1.83(m,2h),1.09(t,j=7.6hz,3h),0.92(t,j=7.2hz,3h).
182、lc-ms:(安捷伦6550质谱仪)[m]+=578.2
183、实施例3:化合物e’的合成
184、005-1合成
185、1.反应式
186、
187、2.投料
188、 名称 m.w w.t(g) mmol eq sm1 139.62 5.00 35.81 1 三光气 296.75 11.69 39.39 1.1 四氢呋喃 / 250ml / 50v
189、3.操作
190、投sm1和四氢呋喃,氮气置换3次,冰浴降温至0℃,加入三光气,搅拌10min后去掉冰浴,室温搅拌24h。过滤,滤饼用正己烷淋洗,滤饼45℃减压干燥,得白色固体6.28g。
191、化合物e’的合成
192、1.反应式
193、
194、2.投料
195、
196、
197、3.操作
198、投sm2(2’,3’-异丙叉-5’-脱氧-5-氟胞苷),二氯甲烷,吡啶,氮气置换三次,冰水浴降温。内温5℃时投入005-1,投完后去掉冰浴自然升温至室温反应。反应2h后tlc监控,dcm∶meoh=10∶1(v/v),254nm下,薄层板底部显蓝光点为产品。反应完成后加少量甲醇淬灭酰氯,将体系旋干,剩余固体进行柱层析纯化。得淡黄色固体。
199、1h nmr(瓦里安,400mhz,cd3od):δ1.33(s,3h,c-ch3),1.38(d,3h,ch3),1.53(s,3h,c-ch3),3.28(s,9h,n(ch3)3),3.50(t,1h,o-ch-c),3.77(t,2h,n-ch2-c),3.83(t,2h,o-ch2-c),4.27(m,1h,c-ch-c),4.56(m,1h,o-ch-c),4.97(d,1h,n-ch-o),5.75(d,1h,n-ch=c),8.13(d,1h,nh).
200、lc-ms:(安捷伦6550质谱仪)[m]+=415.1
201、实施例4:化合物h(2020-sxpt-004)的合成
202、1.反应式
203、
204、2.投料
205、
206、
207、3.操作
208、将乙腈,sm3,sm4,dcc,dmap投入三口瓶中,氮气置换三次,体系呈黄色浊液,加入diea,室温搅拌24h。tlc检测,dcm∶meoh=10∶1(v∶v),sm3反应完,将反应液直接旋干,柱层析纯化得白色膏状物。
209、1h nmr(瓦里安,400mhz,cd3od):δ1.90(m,2h,c-ch2-c),2.38(t,2h,ch2-co),2.56(t,2h,phch2),3.20(s,9h,n(ch3)3),3.62-3.74(m,10h,ch2),4.51(t,2h,coo-ch2),6.68(d,2h,phh),7.06(d,2h,phh).
210、lc-ms:(安捷伦6550质谱仪)[m]+=389.2
211、试验例1药理试验
212、试验方法:将小鼠结肠癌mc38肿瘤细胞悬液以4.0×106个/0.2ml分别接种于小鼠左侧腋背部皮下,接种当天记为d0。d1时,随机分为:溶剂对照组(生理盐水),e1s4955921.25mg/kg,42.5mg/kg(与伊立替康40.0mg/kg等摩尔浓度),64.0mg/kg剂量组、伊立替康40.0mg/kg剂量组共5组,每组为10只动物;分组当天,所有组别动物分别开始腹腔注射给药,共给药3次。
213、实验结束时,眼眶取血20μl,颈椎脱位处死动物,进行血常规检查,剥取肿瘤、胸腺、脾脏并称重,对肿瘤组织进行拍照,根据重量分别计算瘤重抑制率irtw%,胸腺指数、脾指数。
214、试验结果:
215、实验动物在给药期间,除腹腔注射e1s49559 64.0mg/kg剂量组外,荷瘤小鼠体重均有一定程度的上升,但与溶剂对照组的小鼠体重比较,腹腔注射e1s49559 42.5mg/kg、64.0mg/kg和伊立替康40.0mg/kg剂量组的小鼠体重均有不同程度的降低,且具有统计学意义。
216、溶剂对照组肿瘤生长良好,与溶剂对照组比较,e1s49559 21.25mg/kg、42.5mg/kg、64.0mg/kg剂量组对小鼠mc38结肠癌的生长抑制率分别为3.80%、34.62%、51.09%,均小于60%;伊立替康对小鼠mc38结肠癌的生长抑制率为47.26%,小于60%。
217、腹腔注射e1s49559的三个剂量组及伊立替康组的荷瘤小鼠外周血白细胞计数均低于溶剂对照组,但均不具有统计学意义;且上述腹腔注射给药组的荷瘤小鼠外周血单核细胞比率均高于溶剂对照组,其中e1s49559 42.5mg/kg和64.0mg/kg剂量组的单核细胞比率与溶剂对照组比较,差异具有统计学意义。腹腔注射e1s49559 21.25mg/kg剂量组的荷瘤小鼠外周血红细胞计数和血红蛋白量均低于溶剂对照组,且具有统计学意义。腹腔注射e1s49559 42.5mg/kg和64.0mg/kg剂量组的荷瘤小鼠外周血中血小板计数和血小板比积均高于溶剂对照组,且具有统计学意义。
218、溶剂对照组荷瘤小鼠的脾指数为65.36;腹腔注射e1s49559 21.25mg/kg的荷瘤小鼠脾指数为74.91,略高于溶剂对照组;腹腔注射e1s49559 42.5mg/kg和64.0mg/kg的荷瘤小鼠脾指数分别为63.93和55.87,略低于溶剂对照组;阳性对照伊立替康40.0mg/kg的荷瘤小鼠脾指数为53.92,略低于溶剂对照组;但均未通过统计学检验。溶剂对照组荷瘤小鼠的胸腺指数为24.26;腹腔注射e1s49559 21.25mg/kg,42.5mg/kg和64.0mg/kg的荷瘤小鼠胸腺指数分别为20.44,19.02,19.28,其中42.5mg/kg和64.0mg/kg剂量组的胸腺指数明显低于溶剂对照组,且具有统计学意义;腹腔注射伊立替康40.0mg/kg的荷瘤小鼠胸腺指数为13.69,显著低于溶剂对照组,且具有统计学意义。
219、试验结论:
220、腹腔注射伊立替康及e1s49559均可降低荷瘤小鼠的胸腺指数,但等摩尔浓度下(e1s49559 42.5mg/kg,伊立替康40.0mg/kg,),e1s49559对荷瘤小鼠胸腺指数的影响小于伊立替康。
221、试验例2化合物e’和h的细胞活性和毒性实验
222、试验方法:以非活性药物2’,3’-异丙叉-5’-脱氧-5-氟胞苷(sm2)和阳性药物5-氟尿嘧啶(5-fu)为对照,测定化合物的抗肿瘤活性ic50。化合物水相溶解配置100mm母液,10倍梯度稀释10mm、1000μm、100μm、10μm、1μm、0.1μm、10nm等不同浓度。
223、细胞培养及铺板:肿瘤细胞均使用含10%胎牛血清(fbs)的dmem培养基培养,正常肝细胞用含10%fbs及胰岛素的f12k培养基培养,待培养至75-85%密度,消化计数,均匀铺至96孔板。初始细胞数如下表所示:
224、 细胞 每孔细胞数 细胞 每孔细胞数 a549 2000 hct8 4000 hs683 3000 hct8-5fu 4000 hepg2 2000 aml12 4000 hgc27 2000
225、加药处理:细胞铺板24小时后加入待测化合物,每板均设溶剂对照孔。
226、mtt检测:加药72小时后,加入0.5mg/ml mtt,孵育4小时后,弃上清,加100μldmso,摇床水平震荡10min后酶标仪检测od570与od720数值,细胞存活率计算公式:(加药组od570-加药组od720)/(溶剂组od570溶剂组od720)。采用prism graphpad计算ic50,评价各待测化合物体外细胞毒效应。
227、试验结果:测得的化合物ic50见下表
228、
229、各化合物浓度-细胞活性曲线如图5所示。
230、试验结论:化合物e’与其阴性对照药sm2相比,未出现明显的细胞毒性,表明经本发明的水溶结构修饰改造后,没有观察到明显的毒性。化合物h对所有的肿瘤细胞有较好的抑制作用,尤其是对5-氟尿嘧啶的耐药细胞株hct8-5fu的细胞毒作用显著优于5-氟尿嘧啶。上述结果显示本发明涉及的水溶性结构修饰,可以改善部分耐药肿瘤细胞的抑制活性,从而具有良好的应用前景。
231、试验例3,体外药代试验
232、试验方法:
233、大鼠血浆代谢稳定性实验:8组(孵育时间为0,5,15,30min,1,2,4,8h)×2(对照组和bnpp组)×3次平行实验。对照组,孵育体系总体积为100μl,体系包括大鼠血浆93μl+5μl生理盐水,在37℃水浴中预热5min;2μl的候选药物e1s49559(0.1%甲酸水溶液(体积百分比)稀释,加入浓度10μg/ml)加入孵育体系中,涡旋混匀20s,置于37℃水浴中孵育;取样,孵育时间为0,5,15,30min,1,2,4,8h。bnpp组,孵育体系总体积为100μl,体系包括大鼠血浆93μl+5μl bnpp(溶液浓度50mm),涡旋混匀1min,在37℃水浴中预热5min;2μl的候选药物e1s49559(0.1%甲酸水溶液稀释,加入浓度10μg/ml)加入孵育体系中,涡旋混匀20s,置于37℃水浴中孵育;孵育时间为0,5,15,30min,1,2,4,8h。
234、在上述终止反应的样品或标准曲线及质控样品中加入300μl的0.1%甲酸的冰甲醇(体积百分比)(4℃,含内标30ng/ml yltk),涡旋3min,低温离心(14000rpm,5min,4℃),取上清液300μl在室温下采用氮气吹干。取出后加入100μl复溶溶剂(90%乙腈+10%水(体积比)),离心过滤后待lc-ms/ms分析。
235、色谱质谱条件如下:
236、色谱柱:月旭c18:ultimate aq-c18(3.0×50mm,3μm)
237、流动相:a:1mm甲酸铵水溶液,含0.1%甲酸(体积百分比)b:乙腈,进样量:5μl
238、线性梯度洗脱程序如下:
239、
240、采用api 4000串联质谱仪,采用正离子模式的esi检测方式,采集条件如下:喷雾电压(is):5000v;雾化器气(gas1):50psi;辅助气(gas2):60psi;气帘气(cur):30psi;雾化温度(tem):450℃;诱导碰撞气体(cad gas):4;扫描方式为mrm模式,选择q1/q3离子对,进行lc-ms/ms分析。
241、 化合物 dp ce q1 q3 e1s49559 60 40 578.2 99.0 sn-38 80 50 393.1 349.1 yltk 70 45 587.3 124.1
242、实验结果:如图6所示,在e1s49559的esi-ms谱中,观察到m/z 578.24的离子峰,在其二级质谱图中,观察到m/z 519.17和m/z 99.0等离子峰。在标准品sn-38的esi-ms谱中,观察到m/z 393.14的离子峰,在其二级质谱图中,观察到m/z 349.15和m/z 293.12等离子峰。在内标化合物yltk的esi-ms谱中,观察到m/z 587.28的离子峰,在其二级质谱图中,观察到m/z 502.19、m/z 195.14、m/z 167.11、m/z 124.11等离子峰。
243、如图7所示,对照品e1s49559、sn-38和内标yltk在上述lc-ms/ms分析条件下,三者的保留时间分别为0.95,1.12和0.94min。在e1s49559与大鼠血浆孵育后,可以在图7的d中观察到原药e1s49559显著降低,且观察到高含量的sn38。另外,经bnpp羧酸酯酶抑制剂共同孵育后,代谢物sn38的生成明显受到抑制。
244、实验结论:候选药物e1s49559在血浆中可释放活性化合物sn38,表明e1s49559为sn38的前药,且可以经羧酸酯酶介导的水解代谢释放sn38。
245、试验例4,体内药代试验
246、试验方法:
247、大鼠体内代谢实验:
248、试验目的
249、采用lc-ms/hrms方法,对化合物2020-sxpt-004和2020-sxpt-005-2进行药物代谢产物鉴定。
250、仪器与试剂
251、化合物h(2020-sxpt-004)结构如下:
252、
253、化合物e’(2020-sxpt-005-2)结构如下:
254、
255、试验中用到的主要仪器
256、
257、
258、试验中用到的主要试剂
259、
260、实验步骤
261、用生理盐水分别配制5mg/ml的化合物h(2020-sxpt-004)和化合物e’(2020-sxpt-005-2)溶液,按10mg/kg给药剂量各给予3只大鼠,在给药前0min和给药后5min,15min,30min,45min,1h,1.5h,2h,3h,4h,6h,8h,23h不同时间点采集大鼠眼丛静脉血,血样收集到预先冰浴并加有肝素钠的抗凝管中,冰浴条件下进行转运,静置后离心(4000rpm,10min),取上层血浆按100ul分装,置于ep管内,加入300ul体积冰乙腈,离心沉淀蛋白后,取上清-80℃保存备用。
262、各样本以给药前0h为空白对照样品,给药后其余时间点各取一定量混合均匀,将空白样品和给药混合样品在14000rpm离心5min后,取上清液,氮吹挥干溶剂,加100μl复溶溶剂水-乙腈(98∶2 v∶v),涡旋5min,20000rpm离心5min后,取上清液,取10μl进行测定。
263、分别称取一定量2020-sxpt-004和2020-sxpt-005-2置于两个ep管中,用水-乙腈(98∶2 v∶v)溶液稀释至100ng/ml溶液。
264、色谱条件
265、色谱方法主要参数
266、
267、
268、液相梯度洗脱程序:进样前用起始流动相预平衡色谱柱8min梯度洗脱条件
269、
270、质谱条件
271、质谱方法主要参数
272、
273、实验结果
274、利用lc-ms/hrms技术在经静脉注射给药大鼠血浆样本中共鉴定出11个化合物h(2020-sxpt-004)的代谢产物。根据推测的代谢产物结构,化合物h(2020-sxpt-004)在大鼠体内的可能代谢途径如下:
275、
276、利用lc-ms/hrms技术在经静脉注射给药大鼠血浆样本中共鉴定出4个化合物e’(2020-sxpt-005)的代谢产物。根据推测的代谢产物结构,化合物e’(2020-sxpt-005)在体内的可能代谢途径如下:
277、
278、下表汇总了化合物h的体内代谢产物的分子组成、[m+h]+分子量理论值和实测值、误差、样本来源、样本中的峰面积以及相对含量。
279、
280、注:rs为大鼠血浆。
281、下表汇总了化合物e’的体内代谢产物的分子组成、[m+h]+分子量理论值和实测值、误差、样本来源、样本中的峰面积以及相对含量。
282、
283、注:rs为大鼠血浆。
284、实验结论:
285、候选药物h经静脉注射给药后,在大鼠体内中可释放活性化合物苯丁酸氮芥(代谢产物m1),相对含量占6.57%,表明化合物h为苯丁酸氮芥的前药;结合实验例2,化合物h可以作为抗肿瘤药物的使用途径。
286、候选药物e(m0)经静脉注射给药后,在大鼠体内中可主要断裂碳酯键,脱去前药水溶性基团生成代谢产物m1,相对含量占3.44%,符合本发明的水溶性前药设计要求;同时也可以进一步代谢释放活性化合物5-氟尿嘧啶(代谢产物m4),相对含量占0.31%,表明化合物e’为5-氟尿嘧啶的前药,可以作为抗肿瘤药物的使用途径。
287、本技术描述了多个实施例,但是该描述是示例性的,而不是限制性的,并且对于本领域的普通技术人员来说显而易见的是,在本技术所描述的实施例包含的范围内可以有更多的实施例和实现方案。
- 还没有人留言评论。精彩留言会获得点赞!