一种脂溶态砷化合物的合成方法
1.本发明属于有机合成技术领域,涉及一种脂溶态砷化合物的合成方法。
背景技术:
2.脂溶态砷化合物是指细胞内的含砷脂质,包括含砷脂肪酸、含砷碳氢化合物、含砷长链醇、砷糖磷脂等,其结构复杂且含量低,提取难度较高;例如,海洋生物中砷的主要存在形式是脂溶态砷化合物。脂溶态砷化合物的形态包括含脂溶态砷化合物肪酸(arsenic-containing fatty acids,asfas)、含砷碳氢化合物(arsenic-containing hydrocarbons,ashcs)、含砷长链醇、含砷磷脂(arsenic-containing phospholipids,aspls)、含砷磷脂酰胆碱(arsenic-containing phosphatidylcholines,aspcs)和含砷磷脂酰乙醇胺(arsenic-containing phosphati
‑ꢀ
dylethanolamine,aspes)等。
3.此前普遍认知脂溶态砷化合物没有毒性,无需对其进行健康风险评估。《食品安全国家标准食品中污染物限量 gb 2762-2017》中对食品中砷污染物的限量也是以总砷含量和无机砷含量为标准的,对有机砷的含量缺乏关注。现有研究表明脂溶态砷化合物对人类有生物利用性,并被广泛地降解为小的砷化合物。现有研究表明ashcs和asfas具有细胞毒性,且毒性大小ashcs大于asfas,脂溶态砷化合物可以穿过血脑屏障蓄积在脑组织中,造成潜在危害。另外,ashcs会随着哺乳母亲食用后转移至乳汁中,影响婴儿大脑发育,有潜在神经毒性。
4.随着脂溶态砷化合物对人体影响的研究深入,急需商品化可工业化大规模生产的脂溶态砷化合物作为试验原料。但目前国内外并无商品化的脂溶态砷化合物,一般为实验室小规模自行制备。目前脂溶态砷化合物合成方法的流程反应步骤繁多、耗时久,涉及金属钠,如果操作不当很容易存在安全隐患,过程难检测、风险大,而且经常出现产量低且不稳定的情况。
技术实现要素:
5.本发明的目的在于提供一种脂溶态砷化合物的合成方法。所述合成方法具有原料简单、反应简单且耗时短,风险较低,得率稳定、产品纯度高、反应过程易控制、设备要求低的优点,适合进行大规模工业化生产。
6.基于上述目的,本技术通过提供一种脂溶态砷化合物的合成方法来解决该领域中的这种需要。
7.一方面,本发明涉及一种脂溶态砷化合物的合成方法,其包括:在强碱溶液中加入碘化二甲胂得到双(二甲基胂)氧化物;向所述双(二甲基胂)氧化物加入溴代烷烃和无水乙醇反应后得到所述脂溶态砷化合物;所述溴代烷烃包括1-溴十四烷、1-溴十五烷、溴十六烷中的一种。
8.具体地,碘化二甲胂结构如式(1)所示,双(二甲基胂)氧化物结构如式(2)所示,
。
9.进一步地,本发明提供的脂溶态砷化合物的合成方法中,所述碘化二甲胂的制备方法包括:每7mol二甲基胂酸中加入20mol含碘卤化物、5mol亚硫酸氢盐、水和浓盐酸,搅拌反应,加入二氯甲烷和水,混匀后静置分层,收集下层液体,旋蒸浓缩至1ml得到所述碘化二甲胂。
10.具体地,二甲基胂酸结构如式(3)所示,。
11.进一步地,本发明提供的脂溶态砷化合物的合成方法中,所述含碘卤化物为碘化钾,所述亚硫酸氢盐为亚硫酸氢钠;所述搅拌反应的时间不低于4h,所述搅拌反应的速率为800rpm/min;所述浓盐酸的质量分数为32%-37%。优选的,所述浓盐酸的质量分数为32%。
12.进一步地,本发明提供的脂溶态砷化合物的合成方法中,所述强碱溶液为氢氧化钠溶液,所述氢氧化钠溶液的浓度为10mol/l;以摩尔比计,所述强碱溶液的oh-与所述碘化二甲胂的配比为7:25。
13.进一步地,本发明提供的脂溶态砷化合物的合成方法中,所述碘化二甲胂与所述氢氧化钠溶液的反应在无氧环境下进行,反应时间为10-15min,反应温度控制为0℃;优选地,所述无氧环境包括氩气环境或氮气环境。可选地,所述碘化二甲胂与所述氢氧化钠溶液的反应直接在冰水混合浴中进行,转速控制在800rpm/min,满足反应温度控制为0℃的反应条件。
14.进一步地,本发明提供的脂溶态砷化合物的合成方法中,以摩尔比计,所述双(二甲基胂)氧化物、溴代烷烃、无水乙醇的配比为14:5:175。可选地,所述无水乙醇加入后,需要有回流装置。
15.进一步地,本发明提供的脂溶态砷化合物的合成方法中,向所述双(二甲基胂)氧化物加入溴代烷烃和无水乙醇反应后得到所述脂溶态砷化合物;然后进行除杂萃取旋蒸浓缩,所述除杂萃取旋蒸浓缩包括:向所述含脂溶态砷化合物的反应液加水溶解,乙醚除杂,混匀之后静置分层,收集下层溶液,调节ph至中性,加入二氯甲烷萃取,收集下层溶液,旋蒸浓缩,得到所述脂溶态砷化合物的粗品。
16.另一方面,本发明还提供了所述脂溶态砷化合物的粗品纯化方法,所述纯化包括:200-300目的硅胶装柱得到硅胶柱,将所述脂溶态砷化合物的粗品与100-200目的硅胶混合形成拌样硅胶,转移至所述硅胶柱上,用乙酸乙酯与甲醇的混合液梯度洗脱,进行旋蒸氮吹之后得到白色粉末,即为所述脂溶态砷化合物的纯品。
17.进一步地,本发明提供的所述脂溶态砷化合物的粗品纯化方法中,以质量比计,所述脂溶态砷化合物的粗品与所述拌样硅胶的配比为1:1.2-1.5;以质量比计,所述脂溶态砷化合物的粗品与所述硅胶柱的配比为1:20。
18.本发明与现有技术相比具有以下有益效果或者优点:(1)本技术的合成方法步骤简单,其中原料碘化二甲胂和溴代烷烃直接可以购买得到,此外,本技术还提供了利用二甲基砷酸合成碘化二甲胂的方法,二甲基砷酸也同样易于购得。(2)本技术的合成方法安全风险较低,反应原料不包含金属单质钠。避免了金属单质钠十分不稳定,使用不当容易存在安全隐患等问题。(3)本技术的合成方法时间大大缩短,从二甲基砷酸到合成得到脂溶态砷化合物粗品的时间控制在24h以内,且步骤简单。此外,本技术的合成方法所需设备简单,大型仪器只涉及到磁力搅拌器和氮气瓶,其他均为玻璃仪器。(4)本技术的合成方法中涉及的乙醚、二氯甲烷等有机试剂旋蒸浓缩之后可以回收重复利用,减少了废液处理成本,避免造成资源浪费现象。
附图说明
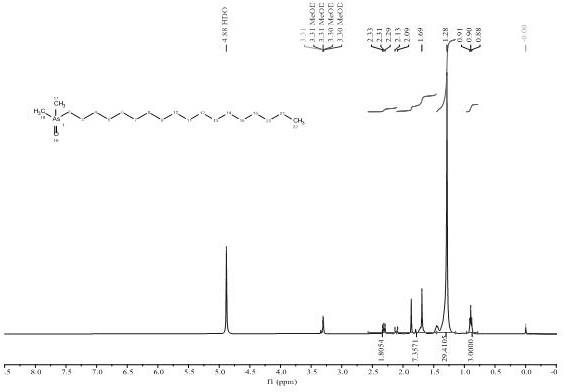
19.图1为实施例1合成得到ashc 374的核磁氢谱图。
20.图2为实施例1合成得到ashc 374的核磁碳谱图。
21.图3为实施例1合成得到ashc 374的高分辨率质谱图。
22.图4为实施例2合成得到ashc 332的核磁氢谱图。
23.图5为实施例2合成得到ashc 332的核磁碳谱图。
24.图6为实施例2合成得到ashc 332的高分辨率质谱图。
25.图7为实施例3合成得到ashc 346的核磁氢谱图。
26.图8为实施例3合成得到ashc 346的核磁碳谱图。
27.图9为实施例3合成得到ashc 346的高分辨率质谱图。
28.其中,ashc 374为相对分子质量为375.2622的含砷碳氢化合物,ashc 332为相对分子质量为332的含砷碳氢化合物,ashc 346为相对分子质量为346的含砷碳氢化合物。
具体实施方式
29.下面,结合实施例对本发明的技术方案进行说明,但是,本发明并不限于下述的实施例。
30.为了使本领域技术人员更好地理解本发明的技术方案能予以实施,下面结合具体实施例和附图对本发明作进一步说明,但所举实施例不作为对本发明的限定。
31.下述各实施例中所述实验方法和检测方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可在市场上购买得到。
32.traar p, rumpler a, madl t, et al.synthesis of naturally occurring arsenic-containing carbohydrates[j].australian journal of chemistry,2009, 62 (6): 538-545.提供了实验室制得脂溶态砷化合物的方法,碘化二甲胂与金属单质钠反应得到二甲基胂化钠溶液,在氩气、0摄氏度加入制得的十五烷基苯磺酸酯(无法市场购得),并在该溶液中加入二甲硫化物,室温下搅拌一夜,回流6h,冷却至室温。将反应混合物在氩气条件下加入乙酸乙酯和水进行萃取,分液,弃去水层,加过氧化氢反应,加盐酸调ph,加乙
酸乙酯萃取,再过阳离子交换柱纯化。该整个合成流程反应步骤繁多、耗时久,涉及金属钠,如果操作不当很容易存在安全隐患,过程难检测、风险大,而且经常出现产量低且不稳定的情况。
[0033]
基于上述现有技术的缺陷,本技术的合成方法步骤简单,其中原料碘化二甲胂和溴代烷烃直接可以购买得到,此外,本技术还提供了利用二甲基砷酸合成碘化二甲胂的方法,二甲基砷酸也同样易于购得;本技术的合成方法安全风险较低,反应原料不包含金属单质钠。避免了金属单质钠十分不稳定,使用不当容易存在安全隐患等问题;本技术的合成方法时间大大缩短,从二甲基砷酸到合成得到脂溶态砷化合物粗品的时间控制在24h以内,且步骤简单。此外,本技术的合成方法所需设备简单,大型仪器只涉及到磁力搅拌器和氮气瓶,其他均为玻璃仪器;本技术的合成方法中涉及的乙醚、二氯甲烷等有机试剂旋蒸浓缩之后可以回收重复利用,减少了废液处理成本,避免造成资源浪费现象。
[0034]
实施例1本实施例提供了脂溶态砷化合物ashc 374的制备方法及过程。
[0035]
ashc 374为相对分子质量为374的含砷碳氢化合物,其结构如下:。
[0036]
称取2 g二甲基胂酸、6.6 g碘化钾、1 g亚硫酸氢钠加入平底三颈烧瓶中,同时加入8 ml水和10 ml 32 %的盐酸,室温下磁力搅拌4 h。加40 ml二氯甲烷和60 ml水,充分混匀直至溶液无颗粒,观察发现溶液由红棕色变成浅黄色,取下层旋蒸浓缩至约1 ml。在氮气和冰水浴条件下,往平底三颈烧瓶加入5 ml10 m氢氧化钠,逐滴加入上述的碘化二甲胂,磁力搅拌15min。再在氮气和78 c水浴条件下,往里加入1.7 g溴代十八烷,10 ml无水乙醇,回流12h。冷却后,加50 ml水和50 ml乙醚,分液后取下层溶液,用6 m hcl调ph为中性。加50 ml二氯甲烷充分萃取,溶液分层,取下层旋蒸浓缩得到粗品。经过硅胶柱分离纯化后,氮吹后可得白色粉末153mg,得率8.2%。
[0037]
对其进行核磁共振分析和质谱分析,得到结果如下:1h nmr (400 mhz, meod) δ 2.1(m, 2h,-as-ch2), 1.7 (s, 6h,-as(ch3)2), 1.6(m, 2h,-ch2),1.2-1.5(m,30h,12-ch2),0.90 (t, j = 6.7 hz, 3h,-ch3);
13
c nmr (101 mhz, meod) δ33.09, 32.94, 31.92, 31.73, 31.58, 30.79, 30.77, 30.75, 30.71, 30.68, 30.52, 30.49, 30.15, 30.10, 23.75, 23.11, 22.85, 14.46, 14.16, 13.61。谱图符合预期ashc 374的结构。同时结合hr-ms分析提示ashc 374相对分子质量为375.2622,与理论值374.253相接近。可以证明该白色粉末为ashc 374,成功合成ashc 374。
[0038]
实施例2本实施例提供了脂溶态砷化合物ashc 332的制备方法及过程。
[0039]
ashc 332为相对分子质量为332的含砷碳氢化合物,其结构如下:
。
[0040]
称取2 g二甲基胂酸、6.6 g碘化钾、1 g亚硫酸氢钠加入平底三颈烧瓶中,同时加入8 ml水和10 ml 32 %的盐酸,室温下磁力搅拌4 h。加40 ml二氯甲烷和60 ml水,充分混匀直至溶液无颗粒,观察发现溶液由红棕色变成浅黄色,取下层旋蒸浓缩至约1 ml。在氮气和冰水浴条件下,往平底三颈烧瓶加入5 ml10 m氢氧化钠,逐滴加入上述的碘化二甲胂,磁力搅拌15min。再在氮气和80 c水浴条件下,往里加入1.45ml 1-溴十五烷,10 ml无水乙醇,回流12h。冷却后,加50 ml水和50 ml乙醚,分液后取下层溶液,用6 m hcl调ph为中性。加50 ml二氯甲烷充分萃取,溶液分层,取下层旋蒸浓缩得到粗品。经过硅胶柱分离纯化后,氮吹后可得白色粉末173mg,得率10.4%。
[0041]
对其进行核磁共振分析和质谱分析,得到结果如下:1h nmr (400 mhz, meod) δ 2.1(m, 2h,-as-ch2), 1.7 (s, 6h,-as(ch3)2), 1.6(m, 2h,-ch2),1.2-1.5(m,24h,12
‑ꢀ
ch2),0.90 (t, j = 6.7 hz, 3h,-ch3);
13
c nmr (101 mhz, meod) δ33.09, 32.94, 31.92, 31.72, 31.58, 30.81, 30.78, 30.53, 30.49, 30.15, 30.10, 23.75, 23.11, 22.85, 14.47, 14.16, 13.60。谱图符合预期ashc 332的结构。同时结合hr-ms分析提示ashc 332相对分子质量为333.2187,与理论值332.206相接近。可以证明该白色粉末为ashc 332,即成功合成ashc 332。
[0042]
实施例3本实施例提供了脂溶态砷化合物ashc 346的制备方法及过程。
[0043]
ashc 346为相对分子质量为346的含砷碳氢化合物,其结构如下:。
[0044]
称取2 g二甲基胂酸、6.6 g碘化钾、1 g亚硫酸氢钠加入平底三颈烧瓶中,同时加入8 ml水和10 ml 32 %的盐酸,室温下磁力搅拌4 h。加40 ml二氯甲烷和60 ml水,充分混匀直至溶液无颗粒,观察发现溶液由红棕色变成浅黄色,取下层旋蒸浓缩至约1 ml。在氮气和冰水浴条件下,往平底三颈烧瓶加入5 ml10 m氢氧化钠,逐滴加入上述的碘化二甲胂,磁力搅拌15min。再在氮气和水浴条件下,往里加入1.58ml 1-溴十六烷,10 ml无水乙醇,回流12h。冷却后,加50 ml水和50 ml乙醚,分液后取下层溶液,用6 m hcl调ph为中性。加50 ml二氯甲烷充分萃取,溶液分层,取下层旋蒸浓缩得到粗品。经过硅胶柱分离纯化后,氮吹后可得白色粉末136 mg,得率7.9%。
[0045]
对其进行核磁共振分析和质谱分析,得到结果如下:1h nmr (400 mhz, meod) δ 2.1(m, 2h,-as-ch2), 1.7 (s, 6h,-as(ch3)2), 1.6(m, 2h,-ch2),1.2-1.5(m,26h,12
‑ꢀ
ch2),0.90 (t, j = 6.7 hz, 3h,-ch3);
13
c nmr (101 mhz, meod) δ33.03, 31.87, 31.74, 30.75, 30.73, 30.71, 30.70, 30.66, 30.48, 30.43, 30.10, 23.70, 23.09, 22.91, 14.44, 13.66。谱图符合预期ashc 346的结构。同时结合hr-ms分析提示ashc 346相对分子质量为347.2304,与理论值346.2217相接近。可以证明该白色粉末为ashc 346,成
功合成ashc 346。
[0046]
如上所述,即可较好地实现本发明,上述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改变和改进,均应落入本发明确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1