一种唑嘧菌胺代谢物的制备方法与流程
1.本发明涉及一种化合物的制备方法,特别涉及一种唑嘧菌胺代谢物的制备方法。
背景技术:
2.唑嘧菌胺(ametoctradin),化学名为:5-ethyl-6-octyl-[1,2,3]triazolo[1,5-a]pyrimidin
–7–
amine,是一种三唑嘧啶类杀菌剂,同时也是一种高选择性的杀菌剂,属于线粒体呼吸抑制剂,对霜霉和疫霉类卵菌纲真菌有控制作用,具有极强的残留活性和耐雨性。经研究确认,唑嘧菌胺具有一种独特的作用模式,唑嘧菌胺可与真菌呼吸复合体中的标桩菌素亚位点结合,从而抑制真菌的活动。这使唑嘧菌胺成为该类别下的唯一一种杀菌剂,与其他商业性杀菌剂无交互抗性,是进行真菌抗性的理想型工具。因此,ametoctradin被许多国家批准用于控制水果和蔬菜的真菌病害。
[0003]
欧洲食品安全局(efsa)在efsa journal 2012;10(11):2921中报道在植物和地下水中发现了高达10.3μg/l的ametoctradin代谢物m650f04(即本发明制备的唑嘧菌胺代谢物)。但目前尚未发现文献报道合成该代谢的方法,因此发明一种唑嘧菌胺代谢物的制备方法对唑嘧菌胺的代谢机理研究有着非常重要的作用。
技术实现要素:
[0004]
发明目的:本发明旨在提供一种高纯度用于唑嘧菌胺的代谢机理研究的唑嘧菌胺代谢物的制备方法。
[0005]
技术方案:本发明所述的种唑嘧菌胺代谢物的制备方法,包括以下步骤:
[0006]
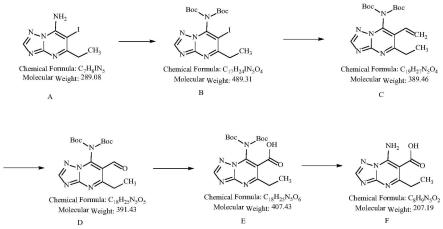
(1)以化合物a 5-乙基-6-碘-[1,2,4]三唑并[1,5-a]嘧啶-7-胺为原料,溶于有机溶剂,加入boc酸酐和碱,反应得到化合物b;
[0007]
(2)将化合物b加入机溶剂中,加入乙烯基锡类化合物和钯催化剂,50~100℃反应,得到化合物c;
[0008]
(3)将化合物c加入有机溶剂中,加入氧化剂,反应将末端碳碳双键氧化为醛,得到化合物d;
[0009]
(4)将化合物d加入有机溶剂中,加入氧化剂和无机碱,反应将末端醛基氧化成酸,得到化合物e;
[0010]
(5)将化合物e加入有机型溶剂中,加入酸,反应去除boc对氨基的保护,得到化合物f,即唑嘧菌胺代谢物;
[0011]
合成路线如下:
[0012][0013]
所述步骤(1)中,采用boc酸酐对化合物a的氨基进行保护。优选的,所述的有机溶剂为四氢呋喃,甲醇,dmf或二氧六环。优选的,所述的碱为三乙胺、n,n-二异丙基乙胺、碳酸钠、碳酸钾或氢氧化钠。优选的,化合物a与boc酸酐的摩尔比为1:1~1:4,化合物a与碱的摩尔比为1:1~1:5。优选的,所述反应温度为20~60℃,所述反应时间为10~20小时;反应过程采用薄层色谱监测反应进程,流动相为正己烷/乙酸乙酯。
[0014]
所述步骤(2)中,化合物a末端i被碳碳双键取代。优选的,所述乙烯基锡类化合物为三丁基乙烯基锡或四乙烯基锡,化合物b与乙烯基锡类化合物的摩尔比为1:1~1:5;优选的,所述钯催化剂为四三苯基膦钯、双三苯基膦二氯化钯,醋酸钯或二(三苯基膦)醋酸钯,化合物b与钯催化剂的摩尔比为1:0.1~1:0.5。优选的,所述有机溶剂为二氯甲烷、1,2-二氯乙烷、氯仿或四氯化碳。优选的,所述反应温度为50~100℃,所述反应时间为10~20小时,反应过程采用薄层色谱监测反应进程,流动相为正己烷/乙酸乙酯。
[0015]
所述步骤(3)中,化合物c的末端碳碳双键被氧化为醛。优选的,所述氧化剂为高锰酸钾、四氧化锇、重铬酸钾、氧气或臭氧,反应温度为-80~-40℃,反应时间为2~10小时,反应过程采用薄层色谱监测反应进程,流动相为二氯甲烷/甲醇。
[0016]
所述步骤(4)中,化合物d的醛基被进一步氧化为酸。优选的,所述的溶剂为正丙醇、异丙醇、正丁醇或叔丁醇。优选的,所述无机碱为碳酸氢钾、磷酸氢二钠、碳酸钠或碳酸氢钠,化合物d与无机碱的摩尔比为1:1~1:5。优选的,所述氧化剂为亚氯酸钠、亚硝酸钠、氯酸钠或硝酸钠,化合物d与氧化剂的摩尔比为1:2~1:10,反应温度为20~60℃,反应时间为1~10小时,反应过程采用薄层色谱监测反应进程,流动相为二氯甲烷/甲醇。
[0017]
所述步骤(5)中,化合物e中被boc保护的氨基脱去boc保护。优选的,所述的酸为盐酸、硫酸、三氟乙酸、醋酸或甲酸,化合物e与酸的摩尔比为1:5~1:50,反应温度为20~60℃,反应0.5~10小时,反应过程采用薄层色谱监测反应进程,流动相为二氯甲烷/甲醇。
[0018]
有益效果:与现有技术相比,本发明具有如下显著优点:该方法通过五步反应制备得到唑嘧菌胺代谢物化合物f,纯度可达99.0%以上,可用于药代动力学研究,为唑嘧菌胺的代谢机理研究提供测试样品,具有重要的应用价值。
附图说明
[0019]
图1为本发明的唑嘧菌胺代谢物的制备方法的合成路线;
[0020]
图2为实施例1步骤(1)产物的质谱图;
[0021]
图3为实施例1步骤(2)产物的质谱图;
[0022]
图4为实施例1步骤(5)产物的质谱图;
[0023]
图5为实施例1步骤(5)产物的氢谱图;
[0024]
图6为实施例1步骤(5)产物的hplc的谱图。
具体实施方式
[0025]
下面结合实施例对本发明的技术方案作进一步说明。
[0026]
实施例1
[0027]
本发明的唑嘧菌胺代谢物的制备方法,包括以下步骤:
[0028]
(1)取50.00g的5-乙基-6-碘-[1,2,4]三唑并[1,5-a]嘧啶-7-胺和113.25g的boc酸酐在四氢呋喃中,加入95.62g的碳酸钠,混合液在30℃反应15小时,薄层色谱显示反应完全,浓缩反应液除去四氢呋喃,硅胶柱纯化得到61.00g的化合物b,收率72.08%。
[0029][0030]
如图2所示,ms:490.3[m+h]
+
;1h nmr(400mhz,dmso)δ18.67(s,1h),3.16(q,2h),1.31(m,21h)。
[0031]
(2)取60.0g的化合物b溶解在氯仿中,加38.88g的三丁基乙烯基锡和1.72g的醋酸钯,在70℃反应15小时,薄层色谱显示反应完全,反应结束,加水萃取,硅胶柱纯化得到39.20g的化合物c,收率82.08%。
[0032][0033]
如图3所示,ms:390.3[m+h]
+
。
[0034]
(3)取39.00g的化合物c溶解在二氯甲烷中,在-60℃通入臭氧反应4小时,薄层色谱显示反应完全,二甲硫醚淬灭,浓缩至干,硅胶柱纯化得到30.00g的化合物d,收率71.53%。
[0035][0036]
(4)取30.00g的化合物d溶解在正丁醇中,冰浴下加入13.86g的亚氯酸钠和45.98g
的磷酸二氢钠,20℃反应8小时,薄层色谱显示反应完全,浓缩加水萃取,粗产物硅胶柱纯化得到20.00g的化合物e,收率64.12%。
[0037][0038]
(5)取20.00g的化合物e溶解在二氯甲烷中,冰浴下加入84.04g的三氟乙酸,50℃反应1小时,薄层色谱显示反应完全,浓缩至干,粗产物c18柱纯化得到7.50g的化合物e,收率73.74%。
[0039][0040]
如图4所示,ms:208.1[m+h]
+
;如图5所示,1hnmr(400mhz,dmso)δ13.60(br,1h),8.69(br,2h),8.50(s,1h),3.10(q,2h),1.22(t,3h);如图6所示,产品纯度99.59%。
[0041]
实施例2
[0042]
本发明的唑嘧菌胺代谢物的制备方法,包括以下步骤:
[0043]
(1)取40.00g的5-乙基-6-碘-[1,2,4]三唑并[1,5-a]嘧啶-7-胺和90.60g的boc酸酐在四氢呋喃中,加入11.07g的氢氧化钠,混合液在20℃反应12小时,薄层色谱显示反应完全,浓缩反应液除去四氢呋喃,硅胶柱纯化得到45.10g的化合物b,收率66.61%;ms:490.3[m+h]
+
。
[0044][0045]
(2)取45.00g的化合物b溶解在氯仿中,加58.32g的三丁基乙烯基锡和2.58g的双三苯基膦二氯化钯,在50℃反应16小时,薄层色谱显示反应完全,反应结束,加水萃取,硅胶柱纯化得到20.50g的化合物c,收率57.23%;ms:390.3[m+h]
+
。
[0046][0047]
(3)取28.00g的化合物c溶解在二氯甲烷中,在-80℃通入臭氧反应8小时,薄层色谱显示反应完全,二甲硫醚淬灭,浓缩至干,硅胶柱纯化得到21.30g的化合物d,收率75.69%。
[0048][0049]
(4)取20.00g的化合物d溶解在叔丁醇中,冰浴下加入4.62g的亚氯酸钠和16.25g的碳酸钠,40℃反应10小时,薄层色谱显示反应完全,浓缩加水萃取,粗产物硅胶柱纯化得到14.20g的化合物e,收率68.27%。
[0050][0051]
(5)取16.00g的化合物e溶解在二氯甲烷中,冰浴下加入59.72g的浓盐酸,60℃反应4小时,薄层色谱显示反应完全,浓缩至干,粗产物c18柱纯化得到5.00g的化合物e,收率61.42%,ms:208.1[m+h]
+
,1h-nmr同实施例1。
[0052][0053]
实施例3
[0054]
本发明的唑嘧菌胺代谢物的制备方法,包括以下步骤:
[0055]
(1)取30.00g的5-乙基-6-碘-[1,2,4]三唑并[1,5-a]嘧啶-7-胺和67.95g的boc酸酐在四氢呋喃中,加入21.93g的碳酸钠,混合液在60℃反应8小时,薄层色谱显示反应完全,浓缩反应液除去四氢呋喃,硅胶柱纯化得到22.10g的化合物b,收率73.67%。
[0056][0057]
(2)取20.0g的化合物b溶解在氯仿中,加14.60g的三丁基乙烯基锡和0.63g的双三苯基膦二氯化钯,在100℃反应12小时,薄层色谱显示反应完全,反应结束,加水萃取,硅胶柱纯化得到14.20g的化合物c,收率81.09%。
[0058][0059]
(3)取14.00g的化合物c溶解在二氯甲烷中,在-40℃通入臭氧反应4小时,薄层色谱显示反应完全,二甲硫醚淬灭,浓缩至干,硅胶柱纯化得到10.50g的化合物d,收率74.63%。
[0060][0061]
(4)取30.00g的化合物d溶解在正丁醇中,冰浴下加入2.31g的亚氯酸钠和9.2g的磷酸二氢钠,60℃反应8小时,薄层色谱显示反应完全,浓缩加水萃取,粗产物硅胶柱纯化得到6.30g的化合物e,收率60.52%。
[0062][0063]
(5)取6.00g的化合物e溶解在二氯甲烷中,冰浴下加入25.21g的三氟乙酸,20℃反应1小时,薄层色谱显示反应完全,浓缩至干,粗产物c18柱纯化得到7.50g的化合物e,收率73.74%。
[0064]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1