恩替卡韦中间体制备新工艺方法及分析方法与流程
1.本发明涉及一种恩替卡韦中间体制备新工艺方法及分析方法,属于化合物技术领域。
背景技术:
2.恩替卡韦中间体,化学名:(1s,2s,3s,5s)-5-(2-氨基-6-苄氧基-9h-嘌呤-9-基)-3-苄氧基-2-苄氧基甲基环戊醇cas号:142217-77-4。用于生产抗病毒药恩替卡韦。
3.现有技术中的工艺路线为:存在的问题在于:起始原料采用手性硼试剂,不仅价格昂贵,且硼试剂遇空气极易放热自燃;环戊二烯和氢化钠原位制备环戊二烯钠的工序放出大量氢气,反应极易冲料,存在安全隐患;采用硅胶柱柱层析提纯,产生大量的废硅胶,且此法过程缓慢,工序时间长;纯化过程为去除异构体,需要多次结晶,收率低。
4.因此,现有技术的合成方法存在诸多问题,我们采用全新的工艺,替换手性硼试剂,消除隐患;同时采用全新的提纯方法,避免产生大量固体废弃物,同时节约时间成本,降低工时,利于工业化生产。
技术实现要素:
5.本发明目的在于提供一种恩替卡韦中间体制备新工艺方法及分析方法,为达到上述目的,本发明是采用下述技术方案实现的:本发明公开了一种恩替卡韦中间体制备新工艺方法,合成路线如下:具体方法为:(1)环戊二烯钠与苄基氯甲基醚低温搅拌至反应完全,再滴加三氟化硼-thf络合物,升温反应后加入30%双氧水和饱和氢氧化钠水溶液,氧化水解完毕后加亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,得到中间体s01;(2)将中间体s01在催化剂下经叔丁基过氧化氢氧化,反应完全后加入亚硫酸钠水溶液淬灭,水洗萃取分液后脱溶,得到中间体s02;(3)将中间体s02、氢化钠在有机溶剂中反应,再滴加溴化苄反应,反应完全后加乙醇淬灭反应,减压脱溶用乙酸乙酯/水萃取分液,有机相脱溶后得到中间体s03;(4)s03与o-6-苄基鸟嘌呤在催化剂下于有机溶剂中搅拌反应完全,减压脱去dmf,
剩余物经硅胶层析提纯,再重结晶得到成品。
6.进一步的,步骤1中,环戊二烯钠与苄基氯甲基醚的摩尔比为:1:(0.8-2.5)。
7.进一步的,步骤2中,中间体s01与叔丁基过氧化氢的摩尔比为:1:(2.4-3.8)。
8.进一步的,步骤2中,催化剂为乙酰丙酮氧钒。
9.进一步的,步骤3中,反应温度为降温至5℃及以下。
10.进一步的,步骤3中,有机溶剂为四氢呋喃。
11.进一步的,步骤4中,有机溶剂为dmf。
12.进一步的,步骤4中,催化剂为氢化锂。
13.进一步的,步骤4中,洗脱溶剂为二氯甲烷和甲醇。
14.进一步的,步骤4中,重结晶为乙醇重结晶。
15.同时还公开了一种恩替卡韦中间体含量检测方法,具体包括:采用高效液相分析方法,为:色谱柱:innoval ods-3 (250mm
×
4.6mm,5μm);流动相a:水;流动相b:乙腈;流速:1.0ml/min;检测波长:254nm;柱温:35℃样品浓度:0.8mg/ml;进样量:10μl;溶剂:80%乙腈水溶液;洗脱梯度:时间流动相a %流动相b %0973109733010903510904097360973与现有技术相比,本发明所达到的有益效果:我们采用全新的工艺,替换手性硼试剂,消除隐患;同时采用全新的提纯方法,避免产生大量固体废弃物,同时节约时间成本,降低工时,利于工业化生产。
附图说明
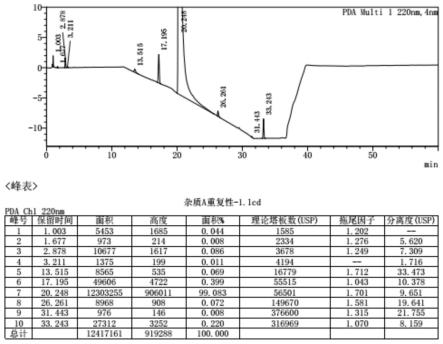
16.图1、本发明的实施例1的中间体s02的液相色谱图;图2、本发明的实施例1的产物的液相色谱图;图3、本发明的实施例2的中间体s02的液相色谱图;图4、本发明的实施例2的产物的液相色谱图;图5、本发明的实施例3的中间体s02的液相色谱图;图6、本发明的实施例3的产物的液相色谱图。
具体实施方式
17.下面对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
18.实施例1反应瓶中加入22g环戊二烯钠,开启搅拌,低温下往其中加入47g苄基氯甲基醚搅拌至反应完全,再往其中滴加制备好的三氟化硼-thf络合物,升温反应后加入25ml 30%双氧水和10ml饱和氢氧化钠水溶液,氧化水解完毕后加25g亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,得到中间体s01共33.26g,产率65.2%。反应瓶中加入34g s01粗品,4.4g乙酰丙酮氧钒,175ml dcm,分批次加入18g叔丁基过氧化氢,用tlc检测反应完全,加入100ml饱和亚硫酸钠水溶液淬灭,静置分液,有机相用100ml水洗萃取分液后脱溶,得到中间体s02共30.7g,产率83.7%。产品纯度为99.08%,见附图所示的液相分析谱图1。
19.反应瓶中加入30.0g s02,300ml四氢呋喃,降温至5℃以下,再加入6.55g氢化钠,搅拌10min后滴加30g溴化苄,tlc检测反应完全后加乙醇淬灭反应,减压脱溶后用乙酸乙酯/水(250ml/250ml)萃取分液,有机相脱溶后得到中间体s03共计30.69g,产率72.6%。
20.反应瓶中加入31g s03,26.55go-6-苄基鸟嘌呤,425mg氢化锂,250ml dmf中搅拌反应,tlc检测反应完全,减压脱去dmf,剩余物经硅胶层析提纯,二氯甲烷和甲醇作为洗脱溶剂,收集产物部分,减压脱溶回收溶剂直接套用,蒸馏产物用100ml乙醇重结晶得到成品
46.06g,产率83.6%。产品纯度为99.94%,见附图所示的液相分析谱图2。
21.实施例2反应瓶中加入22.0g环戊二烯钠,开启搅拌,低温下往其中加入41g苄基氯甲基醚搅拌至反应完全,再往其中滴加制备好的三氟化硼-thf络合物,升温反应后加入30ml 30%双氧水和15ml饱和氢氧化钠水溶液,氧化水解完毕后加20g亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,得到中间体s01共31.02g。
22.反应瓶中加入34g s01粗品,4.1g乙酰丙酮氧钒,200ml dcm,分批次加入15g叔丁基过氧化氢,用tlc检测反应完全,加入100ml饱和亚硫酸钠水溶液淬灭,静置分液,有机相用100ml水洗萃取分液后脱溶,得到中间体s02共25.8g。产品纯度为99.08%,见附图所示的液相分析谱图3。
23.反应瓶中加入30.0g s02,300ml四氢呋喃,降温至5℃以下,再加入6.14g氢化钠,搅拌10min后滴加25g溴化苄,tlc检测反应完全后加乙醇淬灭反应,减压脱溶后用乙酸乙酯/水(250ml/250ml)萃取分液,有机相脱溶后得到中间体s03共计25.82g。
24.反应瓶中加入31g s03,21.59go-6-苄基鸟嘌呤,450mg氢化锂,250ml dmf中搅拌反应,tlc检测反应完全,减压脱去dmf,剩余物经硅胶层析提纯,二氯甲烷和甲醇作为洗脱溶剂,收集产物部分,减压脱溶回收溶剂直接套用,蒸馏产物用100ml乙醇重结晶得到成品42.15g。产品纯度为99.94%,见附图所示的液相分析谱图4。
25.实施例3反应瓶中加入22.0g环戊二烯钠,开启搅拌,低温下往其中加入47g苄基氯甲基醚搅拌至反应完全,再往其中滴加制备好的三氟化硼-thf络合物,升温反应后加入25ml 30%双氧水和10ml饱和氢氧化钠水溶液,氧化水解完毕后加25g亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,得到中间体s01共33.26g,产率65.2%。
26.反应瓶中加入34g s01粗品,4.4g乙酰丙酮氧钒,175ml dcm,分批次加入18g叔丁基过氧化氢,用tlc检测反应完全,加入100ml饱和亚硫酸钠水溶液淬灭,静置分液,有机相用100ml水洗萃取分液后脱溶,得到中间体s02共30.7g,产率83.7%。产品纯度为99.08%,见附图所示的液相分析谱图5。
27.反应瓶中加入30.0g s02,300ml四氢呋喃,降温至5℃以下,再加入6.55g氢化钠,搅拌10min后滴加30g溴化苄,tlc检测反应完全后加乙醇淬灭反应,减压脱溶后用乙酸乙酯/水(250ml/250ml)萃取分液,有机相脱溶后得到中间体s03共计30.69g,产率72.6%。
28.反应瓶中加入31g s03,26.55go-6-苄基鸟嘌呤,425mg氢化锂,250ml dmf中搅拌反应,tlc检测反应完全,减压脱去dmf,剩余物经硅胶层析提纯,二氯甲烷和甲醇作为洗脱溶剂,收集产物部分,减压脱溶回收溶剂直接套用,蒸馏产物用100ml乙醇重结晶得到成品46.06g,产率83.6%。产品纯度为99.94%,见附图所示的液相分析谱图6。
29.实施例4实施例1-3中的液相检测方法为:色谱柱:innoval ods-3 (250mm
×
4.6mm,5μm);流动相a:水;流动相b:乙腈;流速:1.0ml/min;检测波长:254nm;柱温:35℃
样品浓度:0.8mg/ml;进样量:10μl;溶剂:80%乙腈水溶液;洗脱梯度:时间流动相a %流动相b %0973109733010903510904097360973实施例51)反应釜中加入4.40 kg环戊二烯钠,开启搅拌,低温下往其中加入9.40 kg苄基氯甲基醚搅拌至反应完全,再往其中滴加制备好的三氟化硼-thf络合物,升温反应后加入5 l 30%双氧水和2 l饱和氢氧化钠水溶液,氧化水解完毕后加5 kg亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,脱溶物于分子蒸馏脱副产物蒎烯醇,得到中间体s01共6.17 kg,产率60.4%。
30.2)反应釜中加入6.80 kg s01粗品,0.88 kg乙酰丙酮氧钒,35 l dcm,分批次加入3.6 kg叔丁基过氧化氢,用tlc检测反应完全,加入20l饱和亚硫酸钠水溶液淬灭,静置分液,有机相用20l水洗萃取分液后脱溶,得到中间体s02共5.74 kg,产率78.2%。
31.3)反应釜中加入3.0 kg s02,30 l四氢呋喃,降温至5℃以下,再加入0.65kg氢化钠,搅拌10min后滴加2.8 kg溴化苄,tlc检测反应完全后加乙醇淬灭反应,减压脱溶后用乙酸乙酯/水(25l/25l)萃取分液,有机相脱溶后得到中间体s03共计2.78 kg,产率65.8%。
32.4)反应釜中加入3.1 kg s03,2.6 kg o-6-苄基鸟嘌呤,42 g氢化锂,25 l dmf中搅拌反应,tlc检测反应完全,减压脱去dmf,剩余物经硅胶层析提纯,二氯甲烷和甲醇作为洗脱溶剂,收集产物部分,减压脱溶回收洗脱溶液直接套用,蒸馏产物用10l乙醇重结晶得到成品4.31 kg,产率78.2%。
33.实施例61)反应釜中加入4.40 kg环戊二烯钠,开启搅拌,低温下往其中加入10.80 kg苄基氯甲基醚搅拌至反应完全,再往其中滴加制备好的三氟化硼-thf络合物,升温反应后加入5 l 30%双氧水和2 l饱和氢氧化钠水溶液,氧化水解完毕后加5 kg亚硫酸钠淬灭,经水洗萃取分液后,有机相减压脱溶,脱溶物于分子蒸馏脱副产物蒎烯醇,得到中间体s01共6.19kg。
34.2)反应釜中加入6.19 kg s01粗品,0.94kg乙酰丙酮氧钒,35 l dcm,分批次加入3.8kg叔丁基过氧化氢,用tlc检测反应完全,加入20l饱和亚硫酸钠水溶液淬灭,静置分液,有机相用20l水洗萃取分液后脱溶,得到中间体s02共5.47kg。
35.3)反应釜中加入5.47 kg s02,30 l四氢呋喃,降温至5℃以下,再加入0.71kg氢化钠,搅拌10min后滴加3.0 kg溴化苄,tlc检测反应完全后加乙醇淬灭反应,减压脱溶后用乙
酸乙酯/水(25l/25l)萃取分液,有机相脱溶后得到中间体s03共计4.98 kg。
36.4)反应釜中加入4.98 kg s03,4.6 kg o-6-苄基鸟嘌呤,45g氢化锂,25 l dmf中搅拌反应,tlc检测反应完全,减压脱去dmf,剩余物经硅胶层析提纯,二氯甲烷和甲醇作为洗脱溶剂,收集产物部分,减压脱溶回收洗脱溶液直接套用,蒸馏产物用10l乙醇重结晶得到成品5.02kg。
37.以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1