嘧啶并哌啶类FGFR抑制剂及其制备方法和用途与流程
本发明属于药物化学领域,具体而言涉及一类嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物及其制备方法,包含该类化合物的药物组合物,以及该类化合物在制备用于治疗和/或预防fgfr相关性病症、特别是肿瘤的药物中的用途。
背景技术:
1、成纤维细胞生长因子(fifi broblast growth factors,fgfs)结合其受体(fifibroblast growth factor receptors,fgfrs),激活其调控的下游信号通路,在促分裂(胚胎发生、生长发育等)和非促分裂(神经调节、代谢调节等)等生物学过程中起着重要作用。fgfrs是一类典型的受体酪氨酸激酶(receptor tyrosine kinases,rtks),其家族包括fgfr1、fgfr2、fgfr3和fgfr4四种受体,它们均由胞外区、跨膜区和胞内酪氨酸激酶区三个部分组成。其中胞外区包含3个免疫球蛋白样结构(d1-d3),d1区具有自抑制功能,d2和d3区及d2-d3的链接区则与配体结合。fgfr1、fgfr2和fgfr3的ⅲb或ⅲc部分的d3可发生选择性剪接,从而均会产生fgfrb或fgfrc两种亚型,d3域的不同决定了fgfrs的配体结合特异性。fgfs需要在硫酸乙酰肝素糖胺聚糖(heparan sulphate glycosaminoglycan,hsgag)的协助下结合fgfrs,引起了fgfr二聚化,导致其胞内酪氨酸激酶区的多个酪氨酸残基自磷酸化而活化。活化的fgfrs通过磷酸化而激活其底物plcγ和信号衔接蛋白frs2,其底物再激活下游的mek/mapk、pi3k/akt、pkc、stats等信号通路。当内环境紊乱时,fgfr的高表达、突变等导致其信号通路异常激活,与多种疾病的发生发展密切相关。与肿瘤相关的疾病有肺癌、胃癌、乳腺癌、结直肠癌、慢性粒细胞白血病、胆管癌、脑胶质母细胞瘤、软骨肉瘤、脂肪瘤病、膀胱癌等。
2、也与非肿瘤疾病相关,如骨骼疾病(颅缝早闭综合征、kallman综合征、骨质疏松发育不良、软骨发育不良)、性腺发育不全、儿童blaschko线色素减退斑、关节炎。靶向fgfr的抑制剂药物可以抑制fgf/fgfr信号通路的异常激活,具有治疗以上疾病的潜力,fgfr抑制剂药物成为近年来药物研究的热点之一。根据fgfr的蛋白结构,其抑制剂可分为两种类型。第一类是通过靶向膜内酪氨酸激酶域,抑制fgfr的催化活性或酪氨酸自磷酸化,该类抑制剂均为小分子化合物。另一类是靶向fgfr膜外免疫球蛋白域,与fgfs竞争性结合fgfr膜外区,阻断fgf-fgfr结合和通路激活。该类抑制剂有小分子化合物、短肽及抗体。
技术实现思路
1、申请发明人经广泛、深入的研究,通过人工智能(ai)技术手段,设计、合成了一系列结构新颖的小分子化合物,对临床常见的fgfr激酶多种突变体具有很好抑制作用。
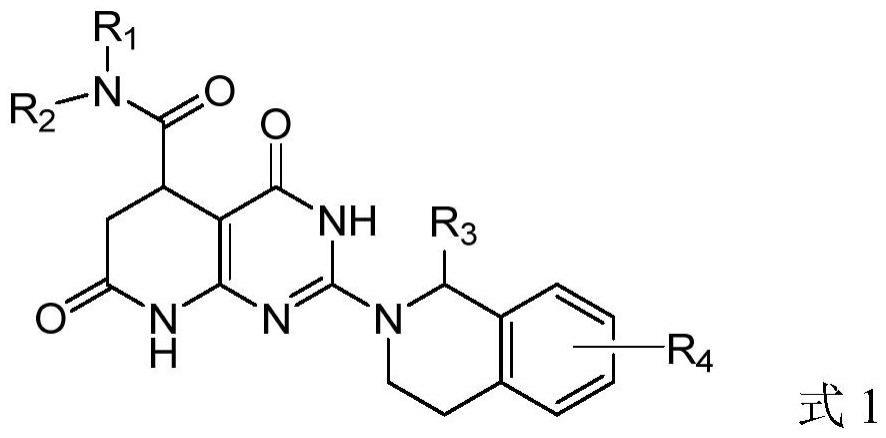
2、根据本发明的一个方面,本发明的一个目的在于提供由以下式1表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物:
3、
4、其中,r1和r2分别各自独立地选自氢原子、羟基、氨基、卤素、氰基、c1至c6的烷基、卤素取代的c1至c6的烷基、c1至c6的烷氧基、卤素取代的c1至c6的烷氧基、取代或未取代的c3至c8环烷基、取代或未取代的c6至c14芳基、含有1-4个选自s、n或o中的杂原子的三至八元取代或未取代的杂芳基、含有1-4个选自s、n或o中的杂原子的三至八元取代或未取代的杂环烷基;在r1和r2中的所述取代或未取代的c3至c8环烷基、取代或未取代的c6至c14芳基、含有1-4个选自s、n或o中的杂原子的三至八元取代或未取代的杂芳基、含有1-4个选自s、n或o中的杂原子的三至八元取代或未取代的杂环烷基中,所述“取代”是指在各个取代基上包含1-3个选自羟基、氨基、卤素、氰基、c1至c6的烷基、卤素取代的c1至c6的烷基、c1至c6的烷氧基、卤素取代的c1至c6的烷氧基的取代基;
5、r3选自氢原子、羟基、氨基、卤素、氰基、c1至c6的烷基、卤素取代的c1至c6的烷基、c1至c6的烷氧基、卤素取代的c1至c6的烷氧基;
6、r4选自氢原子、羟基、氨基、卤素、氰基、c1至c6的烷基、卤素取代的c1至c6的烷基、c1至c6的烷氧基、卤素取代的c1至c6的烷氧基。
7、优选地,r1和r2分别各自独立地选自氢原子、羟基、氨基、卤素、氰基、c1至c3的烷基、卤素取代的c1至c3的烷基、c1至c3的烷氧基、卤素取代的c1至c3的烷氧基、取代或未取代的c3至c6环烷基、取代或未取代的c6至c10芳基、含有1-3个选自n或o中的杂原子的三至六元取代或未取代的杂芳基、含有1-3个选自n或o中的杂原子的三至六元取代或未取代的杂环烷基;在r1和r2中的所述取代或未取代的c3至c6环烷基、取代或未取代的c6至c10芳基、含有1-3个选自n或o中的杂原子的三至六元取代或未取代的杂芳基、含有1-3个选自n或o中的杂原子的三至六元取代或未取代的杂环烷基中,所述“取代”是指在各个取代基上包含1-3个选自羟基、氨基、卤素、氰基、c1至c3的烷基、卤素取代的c1至c3的烷基;
8、优选地,r1和r2分别各自独立地选自氢原子、羟基、氨基、卤素、甲基、乙基、丙基、异丙基、由1-3个氟原子取代的c1至c3烷基、由1-3个氯原子取代的c1至c3烷基、由1-3个溴原子取代的c1至c3烷基、取代或未取代的c5至c6环烷基、取代或未取代的苯基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂芳基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂环烷基;在r1和r2中的所述取代或未取代的c5至c6环烷基、取代或未取代的苯基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂芳基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂环烷基中,所述“取代”是指在各个取代基上包含1-3个选自羟基、卤素、甲基、乙基、丙基、异丙基的取代基。
9、更优选地,当r1和r2分别各自独立地选自取代或未取代的c5至c6环烷基、取代或未取代的苯基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂芳基、含有1或2个选自n或o中的杂原子的五至六元取代或未取代的杂环烷基时,r1和r2具体为选自如下的取代基:
10、
11、优选地,r3选自氢原子、羟基、氨基、卤素、c1至c4的烷基、卤素取代的c1至c4的烷基。
12、优选地,r3选自氢原子、甲基、乙基、丙基、异丙基、由1-3个氟原子取代的c1至c3烷基、由1-3个氯原子取代的c1至c3烷基、由1-3个溴原子取代的c1至c3烷基。
13、优选地,r4选自氢原子、羟基、氨基、卤素、c1至c4的烷基、卤素取代的c1至c4的烷基。
14、优选地,r4选自氢原子、甲基、乙基、丙基、异丙基、由1-3个氟原子取代的c1至c3烷基、由1-3个氯原子取代的c1至c3烷基、由1-3个溴原子取代的c1至c3烷基。
15、优选地,根据本发明的式1表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物由以下式2或式3表示:
16、
17、其中,r1、r2和r3的定义与式1中相同。
18、r5和r6各自独立地选自氢原子、羟基、氨基、卤素、氰基、c1至c6的烷基、卤素取代的c1至c6的烷基、c1至c6的烷氧基、卤素取代的c1至c6的烷氧基。
19、优选地,r5和r6各自独立地选自选自氢原子、羟基、氨基、卤素、c1至c4的烷基、卤素取代的c1至c4的烷基。
20、优选地,r5和r6各自独立地选自选自氢原子、甲基、乙基、丙基、异丙基、由1-3个氟原子取代的c1至c3烷基、由1-3个氯原子取代的c1至c3烷基、由1-3个溴原子取代的c1至c3烷基。
21、根据本发明的所述嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物选自以下化合物中:
22、
23、
24、根据本发明的一个方面,本发明的一个目的在于提供由式1至式3表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物的制备方法,包括以下步骤:
25、
26、步骤1:2-甲硫基-4-氨基-6-羟基嘧啶(化合物1)与丙二酸环(亚)异丙酯(化合物1a)及乙醛酸甲酯(化合物1b)在甲苯中回流反应3~5小时,得到2-(甲硫基)-4,7-二氧代-3,4,5,6,7,8-六氢吡啶并[2,3-d]嘧啶-5-羧酸甲酯(中间体2);
27、
28、步骤2:中间体2在氧化剂作用下,甲硫基氧化,得到2-(甲基磺酰基)-4,7-二氧代-3,4,5,6,7,8-六氢吡啶并[2,3-d]嘧啶-5-羧酸甲酯(中间体6),其中所述氧化剂选自高锰酸钾,重铬酸钾,间氯过氧苯甲酸,高碘酸,过氧苯甲酸,过氧乙酸,过氧化氢异丙苯,过氧化环己酮;
29、
30、步骤3:中间体6与不同的取代基的四氢异喹啉(化合物2a)发生取代反应,得中间体7a;
31、
32、步骤4:中间体7a在氢氧化锂等碱催化下酯解,得到中间体4a;
33、
34、步骤5:带有不同基团的伯胺(nhr1r2)与中间体4a及n,n-二异丙基乙胺(dipea)及2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(hatu)在dmf等有机溶剂中室温反应3~5小时,得到式1的目标化合物。
35、以上制备方法中各个反应式中的取代基r1、r2、r3和r4的定义与式1中相同。
36、根据本发明的一个方面,本发明的一个目的在于提供所述由式1至式3表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物在制备治疗肺癌、胃癌、乳腺癌、结直肠癌、慢性粒细胞白血病、胆管癌、脑胶质母细胞瘤、软骨肉瘤、脂肪瘤病、膀胱癌、骨骼疾病(颅缝早闭综合征、kallman综合征、骨质疏松发育不良、软骨发育不良)、性腺发育不全、儿童blaschko线色素减退斑、关节炎等疾病或病症的药物中的用途。
37、根据本发明的一个方面,本发明的一个目的在于提供一种药物组合物,其包含治疗有效量的根据本发明的由式1至式3表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物和药学上可接受的赋形剂。
38、根据本发明的一个方面,本发明的一个目的在于提供一种用于在有需要的受试者中治疗和/或预防与fgfr激活突变和/或抗药性突变介导相关的疾病的方法,所述方法包括向受试者施用治疗有效量的本发明所述由式1至式3表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物或包含其的所述药物组合物。所述与fgfr激活突变和/或抗药性突变介导相关的疾病包括肺癌、胃癌、乳腺癌、结直肠癌、慢性粒细胞白血病、胆管癌、脑胶质母细胞瘤、软骨肉瘤、脂肪瘤病、膀胱癌、骨骼疾病(颅缝早闭综合征、kallman综合征、骨质疏松发育不良、软骨发育不良)、性腺发育不全、儿童blaschko线色素减退斑、关节炎等疾病或病症。
39、有益效果
40、本发明的由式1至式3表示的嘧啶并哌啶化合物或者其药学上可接受的盐、立体异构体、前药分子或溶剂合物展示了对fgfr激活突变体或抗性突变体形式(一种或多种)具有高抑制活性。本发明的化合物具有较好的物理化学性质和安全毒性参数。此类化合物在由fgfr激活突变和/或抗药性突变介导的疾病(包括癌症)的治疗会有较好的临床效果。
- 还没有人留言评论。精彩留言会获得点赞!