一种基于PacBio测序筛查工业微生物发酵早期杂菌污染的方法与流程
一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法
技术领域
1.本技术涉及分子生物技术领域,尤其涉及一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法。
背景技术:
2.工业尾气通常以燃烧加热或发电的形式回收利用,而燃烧加热或发电的过程中会排放污染物及粉尘等,因此利用工业尾气作为原料直接转化为液态乙醇或其它有机产品,减少污染物的排放,对保护环境有着积极作用。而这项新兴的生物发酵法使用特定微生物菌体-乙醇梭杆菌,由于乙醇梭杆菌是严格厌氧型微生物,并且所采用的发酵工艺是连续发酵,在连续发酵过程中会使用大量的回用水,而回用水中含有菌体代谢产生的有利于菌种生长的生长因子,因此可以提高菌种的生长活性,达到降本增效的目的。但是如果回用水处理不当会引入外来菌种的污染,尤其是厌氧甲烷菌(methane bacteria),因连续发酵阶段的工艺条件也适合甲烷菌的生长,并且甲烷菌还可以利用乙醇梭杆菌代谢中间产物和最终产物乙酸、乙醇和(h2+co2)等基质通过不同的路径转化为甲烷,而大量产生的甲烷将破环乙醇梭杆菌适宜的生长代谢环境,使得甲烷菌与乙醇梭菌呈现竞争关系,虽然目前在发酵初期染甲烷菌相对容易控制,但是到连续发酵稳定期后,不同种类的产甲烷菌对不同抑制剂的敏感性有着很大的不同,同时甲烷菌对甲烷抑制剂的耐药性不易控制及根除,尤其是在夏季的高温条件下甲烷菌污染的更为严重,使得连续发酵的水体等能源介质均有可能被污染,甲烷菌一旦大量繁殖,乙醇梭杆菌的连续发酵将面临着降产、停产,从而造成严重的经济损失,因此在发酵过程中尽快发现污染源并精准的确锁定污染种属显的尤为重要。
3.目前传统的甲烷菌污染检测方法细菌分离培养、显微镜检查、抗原成分检测、分子生物学基因诊断(荧光定量pcr)、气相色谱仪在线实时检测尾气中甲烷变化量等方法,但是大多需要大量时间检测和操作,且整体的检测灵敏度较低,使得在发酵早期的污染源难以及时发现并有效鉴别和快速诊断,因此如何提供一种快速检测且灵敏度高的筛查发酵早期杂菌污染的方法,是目前亟需解决的技术问题。
技术实现要素:
4.本技术提供了一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,以解决现有技术中针对发酵早期杂菌污染的筛查方式难以兼顾高灵敏度和快速检测的技术问题。
5.第一方面,本技术提供了一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,所述方法包括:
6.提取工业微生物早期发酵的微生物样本dna;
7.采用16s rdna通用引物组对所述微生物样本dna进行第一pcr扩增,后进行基因纯化,得到第一扩增产物的上清液;
8.采用含有标签序列的预设引物组对所述上清液进行第二pcr扩增,得到第二扩增产物;
9.混合所述第二扩增产物,并进行基因纯化,后进行上机预文库的制备,得到混样测序文库;
10.对所述混样测序文库进行pacbio测序,后以16s rdna参考序列数据库对测序结果进行比对,得到比对结果;
11.根据所述比对结果,判断微生物样本中是否含有产甲烷菌;
12.若是,则判定工业微生物早期发酵受到杂菌污染。
13.可选的,所述预设引物组包括seq id no.3所示的27f-01、seq id no.4所示的1492r-01、如seq id no.5所示的27f-02、如seq id no.6所示的1492r-02、如seq id no.7所示的27f-03、如seq id no.8所示的1492r-03、如seq id no.9所示的27f-04、如seq id no.10所示的1492r-04、如seq id no.11所示的27f-05、如seq id no.12所示的1492r-05、如seq id no.13所示的27f-06、如seq id no.14所示的1492r-06、如seq id no.15所示的27f-07、如seq id no.16所示的1492r-07、如seq id no.17所示的27f-08和如seq id no.18所示的1492r-08中的至少一组1492r上游引物和27f下游引物。
14.可选的,所述16s rdna通用引物组包括16s rdna通用上游引物和16s rdna通用下游引物,所述16s rdna通用上游引物的核苷酸序列如seq id no.1所示,所述16s rdna通用下游引物的核苷酸序列如seq id no.2所示。
15.可选的,所述16s rdna通用上游引物和所述16s rdna通用下游引物分别包括磷酸化的5’端。
16.可选的,所述混合所述第二扩增产物,并进行基因纯化,后进行上机预文库的制备,得到混样测序文库,具体包括:
17.混合所述第二扩增产物,并进行基因纯化,得到基因纯化产物;
18.使用多组酶对所述基因纯化产物进行末端补平,后进行接头序列的连接,得到预文库;
19.对所述预文库进行dna损伤的修复,并以核酸外切酶进行未正确连接dna的移除,得到混样测序文库。
20.可选的,所述接头序列如seq id no.19所示。
21.可选的,所述多组酶包括t4 dna连接酶、t4聚合酶和t4多聚核苷酸激酶的混合物。
22.可选的,所述核酸外切酶包括核酸外切酶iii和核酸外切酶vii。
23.可选的,所述预文库的dna含量为480ng~500ng。
24.可选的,所述标签序列包括磷酸化的5nt缓冲序列和16nt标签序列。
25.本技术实施例提供的上述技术方案与现有技术相比具有如下优点:
26.本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,通过先对提取出的微生物样本dna采用16s rdna通用引物组进行第一pcr扩增,从而保证微生物样本dna中扩增出足够的待测样本,再对基因纯化后的样本利用含有标签序列的预设引物组进行第二pcr扩增,实现目的片段的富集的同时避免了pcr扩增偏好性导致测序数据异常,由于16s rdna是细菌系统发育和分类鉴定的指标,经过两次pcr扩增后,可以对16s rdna全长序列进行扩增和检测,基因纯化后的混样测序文库再经过pacbio测序并和
16s rdna参考序列数据库比对,不仅可以减少数据拼接导致的误差,直接得到发酵罐内所含的细菌类型,还能对低比例的发酵早期杂菌污染进行检测,从而提高了检测的灵敏度和检测速度。
附图说明
27.此处的附图被并入说明书中并构成本说明书的一部分,示出了符合本技术的实施例,并与说明书一起用于解释本技术的原理。
28.为了更清楚地说明本技术实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
29.图1为本技术实施例提供的方法的流程示意图;
30.图2为本技术实施例提供的方法的详细流程示意图;
31.图3为本技术实施例提供的方法的实际操作流程示意图;
32.图4为本技术实施例提供的各基因组的pcr产物1%琼脂糖溴化乙锭小孔电泳图;
33.图5为本技术实施例提供的混样测序文库的2100质控图;
34.图6为本技术实施例提供的ccs长度分布图;
35.图7为本技术实施例提供的菌种相对丰度分布柱形图;
36.图8为本技术实施例提供的0.1ng甲烷菌dna测序有效数据对比图;
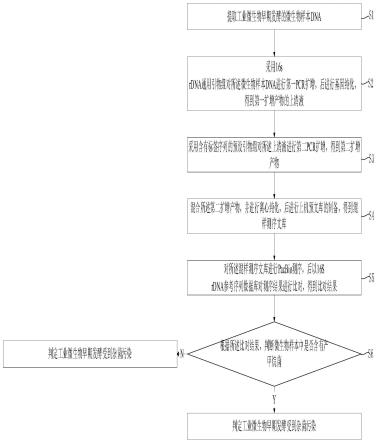
37.图9为本技术实施例提供的极低浓度混入甲烷菌dna测序有效数据比对图。
具体实施方式
38.为使本技术实施例的目的、技术方案和优点更加清楚,下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本技术的一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本技术保护的范围。
39.除非另有特别说明,本技术中用到的各种原材料、试剂、仪器和设备等,均可通过市场购买得到或者可通过现有方法制备得到。
40.本技术的创造性思维为:
41.目前16s rdna基因测序是一种灵敏度高且快速高效检测方法,可以鉴定传统方法无法快速发现的杂菌污染的弊端,同时能够更好的实时了解发酵菌种的状况。由于16s rdna位于原核细胞核糖体小亚基上,其包括10个保守区域和9个高变区域,其中保守区在细菌间差异不大,高变区具有属或种的特异性,随着亲缘关系不同而有一定差异。因此,16s rdna可以作为揭示生物物种的特征核酸序列,被认为最适于细菌系统发育和分类鉴定的指标。利用16s rdna全长测序可对发酵反应体系菌种是否单一进行检测。目前,基于二代高通量测序,随着测序成本的降低,通量增加,检测快捷准确率高,但对于测序要求数据量低的样本来说并不适用,需要攒样本凑数据量,往往会延迟检测周期,这样对早期甲烷菌污染防治有很大影响,导致检测结果与实际发酵罐内情况存在差异,同时二代测序对富含at碱基或gc碱基的区域,若在高度重复序列、回文序列等情况时,会产生gc的较大偏差,使得数据质量不高,针对16s rdna在不同种属之间保守区域相似度大,混合文库上机测序16s rdna
产物拼接上仍存在较大的挑战,而三代测序通量低可快速安排测序,也能规避特殊序列导致的数据偏差,相比与二代测序检测16s rdna v3-v6区域,三代测序读长可达到mbases,一次测序可得到样本16s rdna全长,数据库数据分析结果准确且灵敏性高。由于三代测序具有实时,快速的特点可在4~5h内完成测序流程及数据分析,实时得到序列信息进行物种分类鉴定,完成微生物的快速鉴定,满足动态检测宏基因组需求。
42.因此可以利用pacbio测序方法检测16s rdna全长序列,从而筛查工业微生物早期发酵阶段杂菌污染的情况。
43.如图1所示,本技术实施例提供了一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,所述方法包括:
44.s1.提取工业微生物早期发酵的微生物样本dna;
45.s2.采用16s rdna通用引物组对所述微生物样本dna进行第一pcr扩增,后进行基因纯化,得到第一扩增产物的上清液;
46.s3.采用含有标签序列的预设引物组对所述上清液进行第二pcr扩增,得到第二扩增产物;
47.s4.混合所述第二扩增产物,并进行基因纯化,后进行上机预文库的制备,得到混样测序文库;
48.s5.对所述混样测序文库进行pacbio测序,后以16s rdna参考序列数据库对测序结果进行比对,得到比对结果;
49.s6.根据所述比对结果,判断微生物样本中是否含有产甲烷菌;
50.若是,则判定工业微生物早期发酵受到杂菌污染;
51.若否,则判定工业微生物早期发酵没有受到杂菌污染。
52.本技术实施例中,提取工业微生物早期发酵的微生物样本dna的方式可以选择天根细菌基因组dna提取试剂盒(dp302)提取,具体的提取试剂盒的选择可以根据dna提取纯度片段大小等要求进行,同时工业微生物早期发酵的微生物样本可以来源于发酵罐内的不同时间段的一个或多个微生物样本dna。
53.第一扩增和第二扩增均可按照常规的pcr扩增方法进行扩增,对于具体的dna扩增,本领域技术人员可以依据扩增的核酸、引物及其他条件选择pcr扩增条件。.
54.基因纯化所需的纯化试剂可以采用0.6*ampure pb beads,纯化的方式可以采用切胶柱纯化和blue pippine片段等,可以通过根据片段大小,所需浓度不同选择合适的不同纯化方式。
55.比对的方法可以是软件完成,例如:blast,out和smrt anallysis version2.3等软件,也可以是人工比对的方式逐一比对。
56.在一些可选的实施方式中,所述预设引物组包括seq id no.3所示的27f-01、seq id no.4所示的1492r-01、如seq id no.5所示的27f-02、如seq id no.6所示的1492r-02、如seq id no.7所示的27f-03、如seq id no.8所示的1492r-03、如seq id no.9所示的27f-04、如seq id no.10所示的1492r-04、如seq id no.11所示的27f-05、如seq id no.12所示的1492r-05、如seq id no.13所示的27f-06、如seq id no.14所示的1492r-06、如seq id no.15所示的27f-07、如seq id no.16所示的1492r-07、如seq id no.17所示的27f-08和如seq id no.18所示的1492r-08中的至少一组1492r上游引物和27f下游引物。
57.本技术实施例中,限定预设引物组的具体包括的引物和对应的核苷酸序列,通过这8对特异性引物,可以按照碱基编辑距离8计算获得且进验证可实现数据拆分的目的,从而可以特异性的对微生物样本dna中的16s rdna相关基因进行扩增,提高后续pacbio测序的准确性。
58.同时该预设引物组中的每对引物中的上游引物1492r和下游引物27f都可以随机组合,共可以呈现出64组特异性引物,可同时构建64个文库,方便后续pacbio测序阶段的上机和后续分析,具有测序通量高、时效快、成本低的特点。
59.上述预设引物组的特异性引物的核苷酸序列不局限于本技术所述,只要遵循测序引物设计原则的引物都可以在本技术的方法中使用。
60.在一些可选的实施方式中,所述16s rdna通用引物组包括16s rdna通用上游引物和16s rdna通用下游引物,所述16s rdna通用上游引物的核苷酸序列如seq id no.1所示,所述16s rdna通用下游引物的核苷酸序列如seq id no.2所示。
61.本技术实施例中,限定16s rdna通用引物组的具体核苷酸序列,能保证16s rdna通用引物组对微生物样本dna进行初步扩增,保证后续特异性引物组扩增时,有效的对目的片段进行富集。
62.在一些可选的实施方式中,所述16s rdna通用上游引物和所述16s rdna通用下游引物分别包括磷酸化的5’端。
63.本技术实施例中,限定16s rdna通用引物组的5’端都为磷酸化,保证16s rdna通用引物组结合的准确性,从而保证扩增出样本的数量足够,方便后续的第二pcr扩增的进行,避免pcr扩增偏好性导致测序数据异常,提高pacbio测序阶段的准确性。
64.在一些可选的实施方式中,如图2所示,所述混合所述第二扩增产物,并进行基因纯化,后进行上机预文库的制备,得到混样测序文库,具体包括:
65.s401.混合所述第二扩增产物,并进行基因纯化,得到基因纯化产物;
66.s402.使用多组酶对所述基因纯化产物进行末端补平,后进行接头序列的连接,得到预文库;
67.s403.对所述预文库进行dna损伤的修复,并以核酸外切酶进行未正确连接dna的移除,得到混样测序文库。
68.本技术实施例中,限定具体的上机预文库的制备,通过对第二pcr扩增的扩增产物进行处理,能保证pacbio测序前的混样测序文库中都含有接头序列,并且所有预文库内的dna都是非损伤状态,提高后续pacbio测序的准确性,并提高pacbio测序的灵敏度。
69.上述方法所使用的的dna损伤修复、末端修复连接头及切除未正确连接dna酶都来自neb公司,本技术中也可根据反应条件需求、反应时间长多、酶作用的强度等选用其他公司相关酶进行反应。
70.在一些可选的实施方式中,所述接头序列如seq id no.19所示。
71.本技术实施例中,限定具体接头序列的核苷酸序列,由于接头序列是与pacbio测序的pacbio smrtbell
tm templates相结合的序列,因此引入接头序列,能有效的提高pacbio测序的准确性和灵敏度。
72.在一些可选的实施方式中,所述多组酶包括t4 dna连接酶、t4聚合酶和t4多聚核苷酸激酶的混合物。
73.本技术实施例中,限定多组酶的具体组成,利用t4 dna连接酶、t4聚合酶和t4多聚核苷酸激酶对纯化的第二扩增产物的dna进行末端补平,方便后续接头序列的连接,从而保证pacbio测序的准确性。
74.在一些可选的实施方式中,所述核酸外切酶包括核酸外切酶iii和核酸外切酶vii。
75.本技术实施例中,限定核酸外切酶的具体组成,能保证对未正确连接的dna片段进行移除,从而保证pacbio测序的准确性,同时提高pacbio测序的灵敏度。
76.在一些可选的实施方式中,所述预文库的dna含量为480ng~500ng。
77.在一些可选的实施方式中,所述标签序列包括磷酸化的5nt缓冲序列和16nt标签序列。
78.本技术实施例中,限定标签序列的具体组成,磷酸化的5nt缓冲序列能提高预设引物组的特异性,而16nt标签序列能提高pacbio测序的准确性和灵敏度,从而能够保证pacbio测序的快速检测和高灵敏度。
79.下面结合具体的实施例,进一步阐述本技术。应理解,这些实施例仅用于说明本技术而不用于限制本技术的范围。下列实施例中未注明具体条件的实验方法,通常按照国家标准测定。若没有相应的国家标准,则按照通用的国际标准、常规条件、或按照制造厂商所建议的条件进行。
80.实施例1
81.如图3所示,提供一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,包括:
82.1.使用天根细菌基因组dna提取试剂盒(dp302)提取发酵罐内不同时间段一个或多个微生物样本dna。
83.2.取10ng的dna使用16s rdna的通用引物组对微生物样本dna进行第一pcr扩增,实现对16s rdna全长的扩增,16s rdna的通用引物组的序列是:
84.16s-27f:/5phos/agrgttygatymtggctcag(seq id no.2)
85.16s-1492r:/5phos/rgytaccttgttacgactt(seq id no.1)
86.3.使用0.6*ampure pb beads对产物进行纯化,上清液转移至低吸附pcr管进行第二pcr扩增备用;
87.4.取10ng的第二pcr扩增产物使用含有标签序列的预设引物组(barcoded universal primers)进行第二次pcr扩增,引物如下:
[0088][0089]
5.制备上机预文库:将一个微生物样本或多个微生物样本的第二次pcr扩增产物等量混合,并使用0.6*ampure pb beads纯化,保证上机测序d预文库dna量为480ng~500ng。并使用neb公司的t4 dna连接酶、t4聚合酶,t4多聚核苷酸激酶对预文库dna进行末端补平,然后连接接头序列。其中,接头序列为:
[0090]
/5phos/atctctctcttttcctcctcctccgttgttgttgttgagagagat(seq id no.19)
[0091]
6.使用neb公司的precr修复混合液(precr repair mix)对上机预文库dna损伤进行修复。
[0092]
7.对上机预文库使用neb公司的核酸外切酶iii和核酸外切酶vii移除未正确连接的dna片段,即可得到基于pacbio测序平台的扩增子混样测序文库。
[0093]
8.将混样测序文库使用pacbio dna聚合酶试剂盒退火结合到smrtbell
tm templates,进行测序。
[0094]
9.通过与已知厌氧细菌的16s rdna参考序列数据库比对,确定每个样本中存在的
物种类别。
[0095]
第一pcr扩增所用的pcr反应体系如表1所示。
[0096]
表1第一扩增所用的pcr反应体系情况表
[0097]
模版dna10ngprimermix(10μm/μl each)1.5μlkapa hifi hot start mastermix25μlfinal volumeup to 50μl
[0098]
第一pcr扩增所用的反应条件为:95℃*8min;95℃*30s,57℃*30s,72℃*60s,12cycles;72℃*5min;4℃hold。
[0099]
第二pcr扩增所用的pcr反应体系如表2所示。
[0100]
表2第二扩增所用的pcr反应体系情况表
[0101]
pcr dna10ng2μm barcoded universal primers7μlkapa hifi hot start mastermix25μlfinal volumeup to 50μl
[0102]
第二pcr扩增所用的反应条件为:95℃*8min;95℃*30s,57℃*30s,72℃*60s,8cycles;72℃*5min;4℃hold。
[0103]
接头序列的连接过程所用的pcr反应体系如表3所示。
[0104]
表3接头序列的连接过程所用的pcr反应体系情况表
[0105]
amplicon dna500ng10
×
t4 dna ligase buffer4μlatp(100mm)0.5μldntp(10mm each)0.5μlt4 dna polymerase4μlt4 pnk4μlbg barcode adapter(20μm)20μlenzymatics t4 dna ligase2.5μltotal volume41.5μl
[0106]
接头序列的连接过程的反应条件为:37℃*20min;25℃*15min(lid off);65℃*10min;4℃hold(pcr盖顶温度80℃)
[0107]
dna损伤的修复的过程所用的pcr反应体系如表4所示。
[0108]
表4dna损伤的修复的过程所用的pcr反应体系
[0109]
dna from last step41.5μl10x thermopol reaction buffer5μlnad+(100
×
)0.5μlatp(100mm)0.5μldntp(2.5mm each)0.5μlprecr repair mix2μl
total volume50μl
[0110]
dna损伤的修复的过程的反应条件为:37℃*60min;4℃*1min;4℃hold(pcr盖顶温度42℃)
[0111]
未正确连接dna的移除的pcr反应体系如表5所示。
[0112]
表5未正确连接dna的移除的pcr反应体系情况表
[0113]
ligated dna50μlneb exonucleaseiii0.5μlneb exonucleasevii0.5μltotal volume51μl
[0114]
未正确连接dna的移除的反应条件:37℃*1h,4℃hold(pcr盖顶温度42℃)
[0115]
pacbio测序按照说明书进行。
[0116]
实施例2
[0117]
将实施例2和实施例1进行对比,实施例2和实施例1的区别在于:
[0118]
1.样本dna信息:
[0119]
获取发酵第5天(d5)的样本进行dna提取,其中,所有样本均进行2次重复性测试。对提取出来的甲烷菌基因组进行梯度稀释,并按表6所示的条件混入至d5的基因组样本中并采用实施例1的方法进行测试。
[0120]
表6不同微生物样本的混入d5的基因组样本情况表
[0121]
[0122][0123]
2.上述基因组的pcr产物1%琼脂糖溴化乙锭小孔电泳图(条件:120v,40min)
[0124]
如图4所示,从左到右的点样顺序为:1kb marker、100bp marker、s1、s2、s3、s4、s5、s6。
[0125]
结果显示单个样本pcr扩增产物单一条带,无dimer,混样测序文库的大小1.5kb。
[0126]
3.混样测序文库的2100质控图如图5所示,结果显示上机预文库的插入片段大小集中在1.5kb。
[0127]
4.下机质控数据如表7所示,结合如图6所示的ccs长度分布图可知,每个样本下机数据量稳定,充分说明通过标签引物可以有效的拆分数据,以满足数据分析;同时如图6所示,样本有效数据的序列长度与混样测序文库的构建长度相同。
[0128]
表7下机质控数据情况表
[0129]
samples1s2s3s4s5s6reads of insert330283300633509331193302133014read bases of insert4662063346625223465986924679972467312046635112mean read length of insert153815261538153315381541mean read quality of insert0.99680.99680.99670.99680.99670.9968mean number of passes181818181818
[0130]
5.针对测序数据使用贝叶斯算法,将out与silva数据库进行比对注释,根据属的注释结果,选取在属分类水平上最大相对丰度前5名的属,生成的菌种相对丰度分布柱形图,结果如图7所示,即使在甲烷菌基因组加入量极低时,三代测序技术仍能检测到。
[0131]
6.使用二代高通量测序技术对16s rdna可变区v4-v6区域pcr扩增产物测序,并与16s rdna pacbio测序比较,结果如图8所示,两种测序方式均能检测到相关菌种;同时进行极低浓度混入甲烷菌dna测序有效数据比对,结果如图8所示,16s rdna pacbio全长测序检测灵敏性更高。
[0132]
如图7和图8所示,本技术的混样测序文库构建的方法与常规方法的检测结果比较,常规建库检测方法与本技术的方法对微生物dna进行16s rdna测序的结果高度一致,均
可以检测出杂菌污染,且本技术的方法检测周期短,灵敏度高。
[0133]
同时本技术的混样测序文库构建的方法较常规建库检测方法所检测微生物dna进行16s rdna测序的结果存在一定的差异,本技术的混样测序文库的构建和测序方法所得的结果显示,本技术的方法能够检测出低丰度的杂菌信息,为生产发酵早期污染提供更精准数据分析结果,同时能指导工业微生物发酵过程中对早期杂菌的防控。
[0134]
本技术实施例中的一个或多个技术方案,至少还具有如下技术效果或优点:
[0135]
(1)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,利用2次低循环pcr联合扩增,实现目的片段的富集,有效的降低了pcr扩增偏好性导致测序数据异常。
[0136]
(2)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,使用含标签序列的预设引物组中的上游引物r和下游引物f可以随机组合,使得共有64种组合方式,可同时完成64个文库构建,利用多文库混合上机测序及分析,可以达到测序通量高、时效快、成本低的特点。
[0137]
(3)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,使用含标签序列的预设引物组扩增后将各扩增样本混合至一个离心管中再进行纯化,大大地简化实验操作流程,提高建库效率,降低建库成本。
[0138]
(4)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,可以扩增16s rdna全长序列,从而可以检测所有细菌16s rdna全长,经过与现有数据库中菌种的16s rdna序列比对,可以减少数据拼接导致的误差,直接得到发酵罐内所含的细菌类型,继而为工业微生物发酵杂菌污染分析及治理提供更为精准的数据。
[0139]
(5)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,与常规二代测序数据比较,根据检测混合菌种组成分析,两种测序手段均可以筛查出微生物发酵污染杂菌类别及比例,但对于低比例早期杂菌污染测试样本测序数据分析发现,本技术的方法灵敏度高,可以更早发现杂菌污染现象。
[0140]
(6)本技术实施例提供的一种基于pacbio测序筛查工业微生物发酵早期杂菌污染的方法,整体检测周期短,可在4h~5h内完成测序流程及数据分析,快速得到序列信息进行物种分类鉴定,并且可以完成微生物的快速鉴定,满足动态检测宏基因组需求。
[0141]
本技术的各种实施例可以以一个范围的形式存在;应当理解,以一范围形式的描述仅仅是因为方便及简洁,不应理解为对本技术范围的硬性限制;因此,应当认为所述的范围描述已经具体公开所有可能的子范围以及该范围内的单一数值。例如,应当认为从1到6的范围描述已经具体公开子范围,例如从1到3,从1到4,从1到5,从2到4,从2到6,从3到6等,以及所述范围内的单一数字,例如1、2、3、4、5及6,此不管范围为何皆适用。另外,每当在本文中指出数值范围,是指包括所指范围内的任何引用的数字(分数或整数)。
[0142]
在本技术中,在未作相反说明的情况下,使用的方位词如“上”和“下”具体为附图中的图面方向。另外,在本技术说明书的描述中,术语“包括”“包含”等是指“包括但不限于”。在本文中,诸如“第一”和“第二”等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。在本文中,“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b的情况。其中a,b可以是单
数或者复数。在本文中,“至少一个”是指一个或者多个,“多个”是指两个或两个以上。“至少一种”、“以下至少一项(个)”或其类似表达,是指的这些项中的任意组合,包括单项(个)或复数项(个)的任意组合。例如,“a,b,或c中的至少一项(个)”,或,“a,b,和c中的至少一项(个)”,均可以表示:a,b,c,a-b(即a和b),a-c,b-c,或a-b-c,其中a,b,c分别可以是单个,也可以是多个。
[0143]
以上所述仅是本技术的具体实施方式,使本领域技术人员能够理解或实现本技术。对这些实施例的多种修改对本领域的技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本技术的精神或范围的情况下,在其它实施例中实现。因此,本技术将不会被限制于本文所示的这些实施例,而是要符合与本文所申请的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1