2,6-二氯苯腈的制备方法与流程
2,6-二氯苯腈的制备方法
技术领域
1.本发明涉及有机合成技术领域,尤其涉及一种2,6-二氯苯腈的制备方法。
背景技术:
2.2,6-二氯苯腈是一种除草剂,对杂草有优秀的抑制作用;同时也是农药、医药、材料中间体,可以用来合成2,6-二氟苯腈、2,6-二氟苯甲酰胺、它虫隆、除虫脲、氟螨脲、草克乐等多种农药。这些都具有高效杀虫、低残留等特点。
3.现有技术中,2,6-二氯苯腈的合成路线有很多,主要有以下几类:
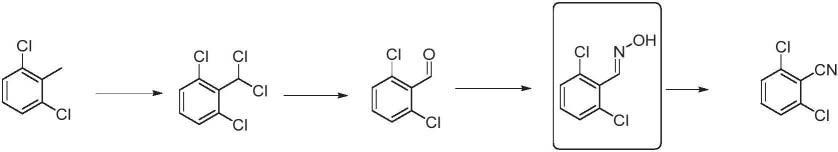
4.a)以2,6-二氯苯甲醛肟为原料或者中间体,在脱水剂作用下,得到2,6-二氯苯腈。但上述反应过程的后处理复杂,需要经过冷却、结晶析出,再经过滤、洗涤、烘干等处理。在中国发明专利cn103382166b和cn103588679.a中,采用的正是这种路线,如下式所示。
[0005][0006]
b)以2,6-二氯苯胺为原料,经过重氮化、氰化反应得到2,6-二氯苯腈。该合成路线使用到了剧毒的氰化物,不适于大量制备,其反应过程如下式所示。
[0007][0008]
c)氨氧化法是目前国际上制备芳香腈类化合物的有效的方法,相关研究较多。
[0009]
在中国发明专利cn1055028c中,首次将2,6-二氯甲苯经过氨氧化制备2,6-二氯苯腈,这是国内第一次报道使用该方法,以钒、钛为主催化剂,以磷、铬为助催化剂的四组分催化剂,高效高选择性地合成了2,6-二氯苯腈。
[0010][0011]
与上述途径类似,在中国发明专利cn102924328a中,采用固定床工艺,以v
1.0
alatibpckdcseof为催化剂,反应收率最高可以达到88%。在中国发明专利cn103041838a中,以硅胶为载体,以钒、铬为主催化剂元素,以钾、锂等为助催化剂元素,制备了2,6-二氯苯腈。相关的专利还有cn103551176a、cn104326940a和cn110404528b。但这些现有技术均使用了复杂且昂贵的多元素或多组分催化剂,催化剂的成本极大。
[0012]
可见已报道的方法,主要有以下缺点:步骤繁琐、反应时间长、温度较高、使用到剧毒试剂、反应副产物多、高温易分解等。
技术实现要素:
[0013]
针对现有技术的不足,本发明的目的在于提供一种一种2,6-二氯苯腈的制备方法。
[0014]
为实现前述发明目的,本发明采用的技术方案包括:
[0015]
第一方面,本发明提供一种2,6-二氯苯腈的制备方法,其包括:
[0016]
使2,6-二氯溴苯与二氧化碳和氨气在催化剂、配体和还原剂的作用下发生取代反应,生成2,6-二氯苯腈;
[0017]
其中,所述催化剂包括铑类催化剂,所述还原剂包括硅烷类还原剂,所述配体包括有机磷配体。
[0018]
基于上述技术方案,与现有技术相比,本发明的有益效果至少包括:
[0019]
本发明提供的一种2,6-二氯苯腈的制备方法以易得的co2和nh3为氰源,通过一步合成即可得到较高产率的2,6-二氯苯腈,具有原料易得、反应步骤少、绿色环保、原料毒性小、易于大量制备的优点。
[0020]
上述说明仅是本发明技术方案的概述,为了能够使本领域技术人员能够更清楚地了解本技术的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例说明如后。
具体实施方式
[0021]
鉴于现有技术中的不足,本案发明人经长期研究和大量实践,得以提出本发明的技术方案。如下将对该技术方案、其实施过程及原理等作进一步的解释说明。
[0022]
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是,本发明还可以采用其他不同于在此描述的方式来实施,因此,本发明的保护范围并不受下面公开的具体实施例的限制。
[0023]
本发明以2,6-二氯溴苯为初始原料,铑为催化剂,有机单膦配体为配体,二氧化碳和液氨为氰源,硅氢烷为还原剂,一步法合成得到2,6-二氯苯腈。该方法环境友好,绿色原子经济性高,产率高,避免剧毒氰源的使用,适合工业化生产。
[0024]
基于此,本发明实施例提供一种2,6-二氯苯腈的制备方法,包括如下的步骤:
[0025]
使2,6-二氯溴苯与二氧化碳和氨气在催化剂、配体和还原剂的作用下发生取代反应,生成2,6-二氯苯腈;
[0026]
其中,所述催化剂包括铑类催化剂,所述还原剂包括硅烷类还原剂,所述配体包括有机磷配体。
[0027]
作为上述技术方案的一些典型的应用示例,上述制备方法可以采用如下的具体步骤得以实施:
[0028][0029]
如上反应式所示,在反应器中,以2,6-二氯溴苯、co2、nh3、还原剂、溶剂、铑类催化剂和配体在一定温度条件下,反应一定时间后即可得到2,6-二氯苯腈。
[0030]
上述技术方案中,二氧化碳和氨气作为氰源,能够在特定的反应条件下被硅烷还
原脱氧生成硅基异氰酸酯,磷配体与铑类催化剂结合形成可溶于溶剂的均相催化剂,并提高铑金属的催化活性,然后2,6-二氯溴苯在特定的催化剂配合物的作用下生成2,6-二氯苯基铑物种,进而进攻硅基异氰酸酯,形成硅基2,6-二氯苯甲酰胺铑,该物种再经硅烷脱氧还原生成2,6-二氯苯腈,并再生催化剂,从而实现不断地催化反应过程。
[0031]
在一些实施方案中,所述铑类催化剂可以包括[rh(cod)c1]2、rh(pph3)3cl、rhi3、[rh(co)2acac]、rh(pph3)3br、[rh(acac)3]、[rhcl(co)(pph3)2]中的任意一种或两种以上的组合;
[0032]
在一些实施方案中,所述配体能够与铑类催化剂结合形成溶于有机溶剂的均相催化剂,具有提高催化剂活性的能力。并且,所述配体优选为有机磷配体,更加优选为具有六元碳环结构(例如苯环或环己烷)或含氮碳杂环的磷的三元衍生物配体。
[0033]
在一些实施方案中,所述配体可以包括具有式l
1-l
12
所示的分子结构的化合物中的任意一种或两种以上的组合:
[0034][0035]
在一些实施方案中,所述硅烷类还原剂可以包括c1sih3、phsih3或etsih3中的任意一种或两种以上的组合。
[0036]
在一些实施方案中,所述取代反应的温度可以为40-180℃,时间可以为3-8小时。
[0037]
在一些实施方案中,所述2,6-二氯溴苯与铑类催化剂的摩尔比可以为1∶0.01-1∶0.2。
[0038]
在一些实施方案中,所述铑类催化剂与有机磷配体的摩尔比可以为1∶2-1∶4。
[0039]
在一些实施方案中,所述2,6-二氯溴苯与硅烷类还原剂的摩尔比可以为1∶1-1∶
10。
[0040]
在一些实施方案中,所述二氧化碳的压力可以为1-10个大气压;
[0041]
在一些实施方案中,所述氨气的压力可以为1-20个大气压。
[0042]
在一些实施方案中,所述制备方法具体可以包括如下的步骤:
[0043]
使所述2,6-二氯溴苯、催化剂、配体和还原剂在有机溶剂中构成液相反应体系,并使所述液相反应体系置于二氧化碳和氨气的混合气氛中进行所述取代反应。
[0044]
在一些实施方案中,所述有机溶剂包括四氢呋喃、2-甲基四氢呋喃、乙腈、乙酸乙酯、甲苯、二甲苯、1,4-二氧六环、二甲亚砜、n,n-二甲基甲酰胺中的任意一种或两种以上的组合。
[0045]
在一些实施方案中,所述制备方法还可以包括如下的步骤:所述取代反应结束后,采用饱和碳酸氢钠水溶液淬灭所述取代反应;
[0046]
在一些实施方案中,所述制备方法还可以包括如下的步骤:对经过所述取代反应后的液相反应体系进行萃取、干燥以及纯化处理,获得2,6-二氯苯腈。
[0047]
以下通过若干实施例进一步详细说明本发明的技术方案。然而,所选的实施例仅用于说明本发明,而不限制本发明的范围。
[0048]
另需说明,如无额外说明,下述实施例中所采用的各种反应原料、催化剂、试剂以及反应装置、提纯处理方法,均是通过常规商购即可获得的或属于常规的实验手段。
[0049]
实施例1
[0050]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0051]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、[rh(cod)cl]2(摩尔比为1%)、配体l1(摩尔比为3.5%)、co2(2atm)、nh3(latm)、c1sih3(摩尔比为1)、四氢呋喃和磁子后,体系升温至70℃,反应8小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用乙酸乙酯萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二氯苯腈,收率56%。
[0052]
本实施例所获产物的核磁数据为:1h nmr(cdcl3,400m hz)δ7.52-7.41(m,3h)ppm;
13
c nmr(cdcl3,100m hz)δ138.4,134.0,128.3,114.3,113.4ppm.
[0053]
实施例2
[0054]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0055]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、rh(pph3)3c1(摩尔比为2%)、配体l3(摩尔比为7%)、co2(1atm)、nh3(2atm)、phsih3(摩尔比为3)、2-甲基四氢呋喃和磁子后,体系升温至90℃,反应8小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用二氯甲烷萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二氯苯腈,收率66%。
[0056]
实施例3
[0057]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0058]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、rhi3(摩尔比为4%)、配体l6(摩尔比为8%)、co2(4atm)、nh3(3atm)、etsih3(摩尔比为5)、甲苯和磁子后,体系升温至100℃,反应6小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用甲苯萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二
氯苯腈,收率71%。
[0059]
实施例4
[0060]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0061]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、[rh(co)2acac](摩尔比为6%)、配体l8(摩尔比为15%)、co2(1atm)、nh3(4atm)、etsih3(摩尔比为7)、二甲苯和磁子后,体系升温至120℃,反应4小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用二氯甲烷萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二氯苯腈,收率76%。
[0062]
实施例5
[0063]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0064]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、rh(pph3)3br(摩尔比为8%)、配体l10(摩尔比为20%)、co2(5atm)、nh3(5atm)、phsih3(摩尔比为8)、1,4-二氧六环和磁子后,体系升温至110℃,反应5小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用二氯甲烷萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二氯苯腈,收率81%。
[0065]
实施例6
[0066]
本实施例示例一2,6-二氯苯腈的制备过程,具体如下所示:
[0067]
向反应器中,加入2,6-二氯溴苯(摩尔比为1)、[rhcl(co)(pph3)2](摩尔比为10%)、配体l12(摩尔比为25%)、co2(2atm)、nh3(6atm)、c1sih3(摩尔比为10)、二甲亚砜和磁子后,体系升温至150℃,反应3小时后冷却,用饱和碳酸氢钠水溶液淬灭反应,用二氯甲烷萃取体系中有机相,合并有机相并用硫酸镁干燥30分钟后,硅藻土过滤。有机相浓缩后,粗产品经重结晶纯化后可得2,6-二氯苯腈,收率90%。
[0068]
对比例1
[0069]
本对比例与实施例2大体相同,区别主要在于:
[0070]
将铑类催化剂替换为钯类催化剂pd(pph3)2cl2。其余原料、试剂的选择以及反应的配比及条件均不变。
[0071]
采用本对比例的合成方法,根本无法合成所期望获得的产物。
[0072]
对比例2
[0073]
本对比例与实施例1大体相同,区别主要在于:
[0074]
将铑类催化剂替换为铱类催化剂[ir(cod)c1]2。其余原料、试剂的选择以及反应的配比及条件均不变。
[0075]
采用本对比例的合成方法,根本无法合成所期望获得的产物。
[0076]
对比例3
[0077]
本对比例与实施例1大体相同,区别主要在于:
[0078]
将有机磷配体中的p元素替换为n元素。其余原料、试剂的选择以及反应的配比及条件均不变。
[0079]
采用本对比例的合成方法,2,6-二氯苯腈的产率极低(<5%)。
[0080]
对比例4
[0081]
本对比例与实施例1大体相同,区别主要在于:
[0082]
将有机磷配体中的p元素替换为as元素。其余原料、试剂的选择以及反应的配比及条件均不变。
[0083]
采用本对比例的合成方法,2,6-二氯苯腈的产率极低(<5%)。
[0084]
对比例5
[0085]
本对比例与实施例1大体相同,区别主要在于:
[0086]
将硅烷类还原剂替换为常见的还原剂氢气。其余原料、试剂的选择以及反应的配比及条件均不变。
[0087]
采用本对比例的合成方法,根本无法合成所期望获得的产物。
[0088]
对比例6
[0089]
本对比例与实施例1大体相同,区别主要在于:
[0090]
将硅烷类还原剂替换为常见的还原剂硼烷。其余原料、试剂的选择以及反应的配比及条件均不变。
[0091]
采用本对比例的合成方法,根本无法合成所期望获得的产物。
[0092]
对比例7
[0093]
本对比例与实施例1大体相同,区别主要在于:
[0094]
将硅烷类还原剂替换为常见的还原剂锌粉。其余原料、试剂的选择以及反应的配比及条件均不变。
[0095]
采用本对比例的合成方法,根本无法合成所期望获得的产物。
[0096]
基于上述检测结果,可以明确,本发明实施例提供的一种2,6-二氯苯腈的制备方法以易得的co2和nh3为氰源,通过一步合成即可得到较高产率的2,6-二氯苯腈,具有原料易得、反应步骤少、绿色环保、原料毒性小、易于大量制备的优点。
[0097]
并且,为实现本发明的技术效果,需要特定的铑类催化剂、硅氢烷类还原剂以及特定的有机磷类配体的三者的组合,上述三者的组合对相关的合成反应的催化作用是具有极大的促进作用的,缺一不可。
[0098]
应当理解,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1