一种2-氯-6-氟三氯苄脱氯还原方法与流程
本发明属于精细化工领域,涉及精细医药中间体的合成和提纯技术,特别是涉及一种工艺简单,产品收率高,降低成本的2-氯-6-氟三氯苄脱氯还原方法。
背景技术:
1、2-氯-6-氟苯甲醛是人用抗生素氟氯西林钠的关键中间体,全球年需求量在1000吨左右。2-氯-6-氟苯甲醛的生产路线主要有两条,第一种是以3-氯-2-甲基苯胺为原料,通过重氮和氟代,制得2-氯-6-氟甲苯,然后再通过光氯化、精馏和水解的工艺路线;第二种是以甲苯为原料,通过氯化和精馏制得2,6-二氯甲苯,然后再通过氟代和精馏,得到2-氯-6-氟甲苯,最后通过光氯化、精馏和水解的工艺路线。
2、两种工艺路线各有其优点,但由于药品对于杂质控制严格,第二种生产路线相关杂质难以控制,而且转化效率也非常低,没有实际的工业化价值,目前国内外均以第一种路线进行生产。
3、2-氯-6-氟甲苯在光氯化过程中,为了控制氯化的转化效率和精馏能耗,在光氯化过程中,约有3%的原料被转化为2-氯-6-氟三氯苄,造成原料损失,并产生大量的副产物无法资源化利用,只能作为危废进行处置。
4、当然也有将2-氯-6-氟三氯苄通过催化水解来制备2-氯-6-氟苯甲酸,但2-氯-6-氟苯甲酸为医药中间体,客户对产品质量提出严格的要求,具体质量指标如下:2-氯-6-氟苯甲酸含量大于99.5%,相关单杂小于0.2%,熔点在156-158℃,残渣小于0.5%。
5、由于生产2-氯-6-氟甲苯的原料3-氯-2-甲基苯胺在生产过程中,其异构体5-氯-2-甲基苯胺很难被彻底除去(目前采用精馏和重结晶相结合的工艺,最低也只能将其控制在0.3%以下),导致制得的2-氟-6-氟甲苯中含有约0.3%的4-氟-2-氟甲苯。但4-氟-2-氟甲苯由于空间位阻比2-氟-6-氟甲苯小,侧链上的甲基更容易被氯化,全部被转化为4-氯-2-氟三氯苄(水解后转化为4-氯-2-氟苯甲酸),虽然对2-氯-6-氟苯甲醛的质量没有影响,但直接影响副产2-氯-6-氟苯甲酸的质量。
6、由于上述原因造成了2-氯-6-氟苯甲醛生产过程中的副产酸杂质含量高(主要杂质为2-氯-6-氟苯甲醛、4-氯-2-氟苯甲酸、焦油、硫酸钠和氯化钠等),由于之前没有合适的精制提纯方法,无法进行商品化应用,大量的副产酸只能暂存在仓库或作为危废进行处置,造成资源浪费和环境污染。
7、之前,对于2-氯-6-氟苯甲酸的精制工艺方面,为了解决目前现有技术中存在的技术难题,发明人也曾尝试过采用升化法、大孔树脂吸附法、酸碱重结晶法、萃取法和甲醇重结晶法等方法进行提纯,但都不成功,或者是收率低,或者是污染严重,或者就是产品质量达不到要求。同时,即使做成高纯度的2-氯-6-氟苯甲酸,市场的需求也非常小。
8、2-氯-6-氟三氯苄是生产2-氯-6-氟苯甲醛生成的副产物,一般含量≥90%,2-氯-6-氟二氯苄含量≤5%,其余为其它杂质。如果能将2-氯-6-氟三氯苄通过还原脱氯的办法,转化为2-氯-6-氟氯苄或2-氯-6-氟二氯苄,那可以实现资源化利用,既降低生产成本,还减少了三废产生量。
技术实现思路
1、为了解决上述问题,本发明提供了一种2-氯-6-氟三氯苄脱氯还原方法,发明人通过大量的实验研究,发现2-氯-6-氟三氯苄在溶剂和催化剂作用下可以跟乙二胺四乙酸二钠盐进行脱氯还原反应,得到2-氯-6-氟二氯苄和2-氯-6-氟一氯苄,具有工艺简单、产品收率高和质量优等特点。
2、为了实现上述目的,本发明采用以下技术方案:
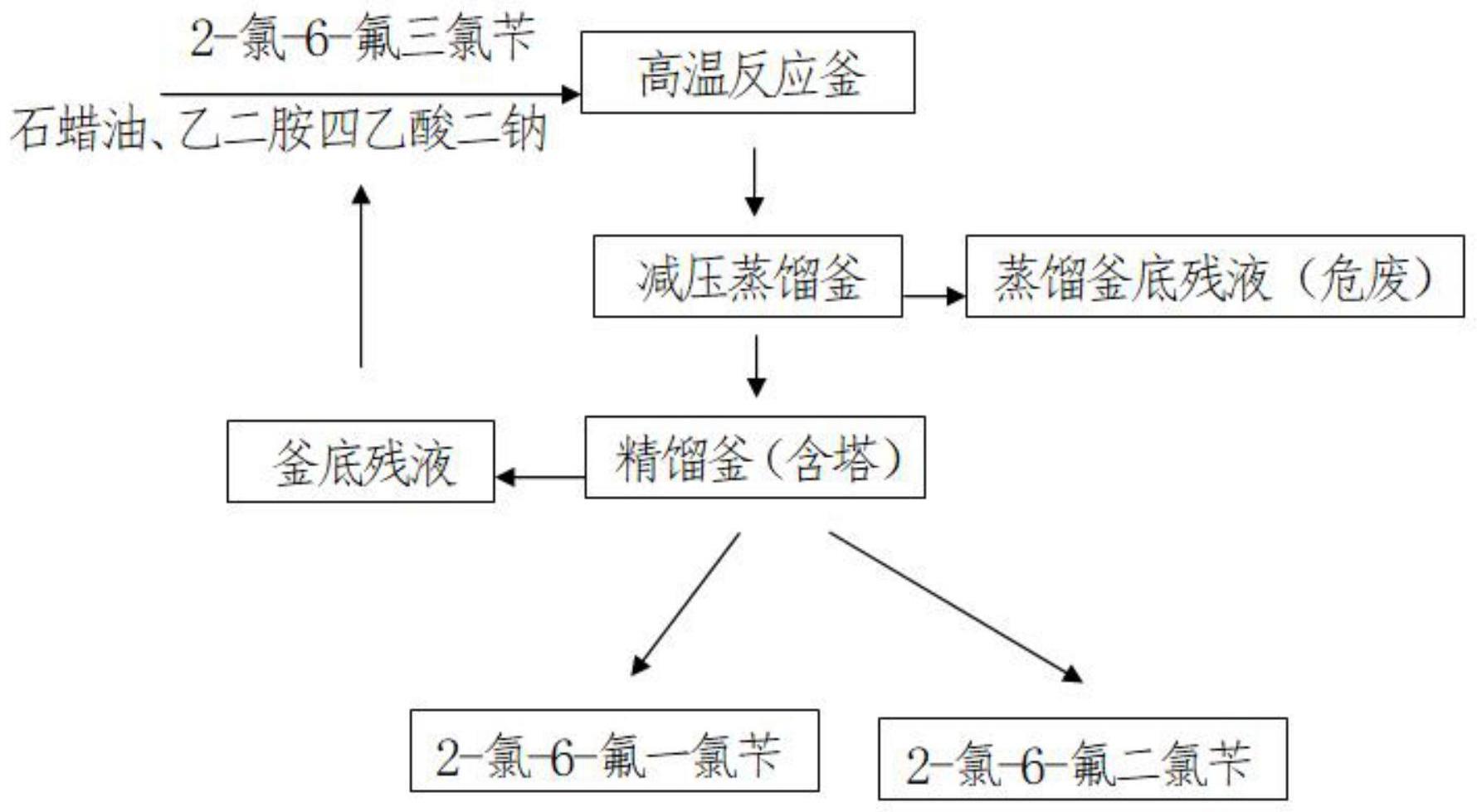
3、本发明提供了一种2-氯-6-氟三氯苄脱氯还原方法,以2-氯-6-氟三氯苄为起始原料,在溶剂和催化剂作用下跟乙二胺四乙酸二钠盐进行脱氯还原反应,得到反应混合物;反应所得混合物通过蒸馏和精馏,分离得到产品2-氯-6-氟二氯苄和2-氯-6-氟一氯苄。
4、作为本发明的一种优选方案,所述2-氯-6-氟三氯苄脱氯还原方法包括以下步骤:
5、1)将作为起始原料的2-氯-6-氟三氯苄加热至融化,然后投入反应釜中;
6、2)向反应釜内投入溶剂、催化剂和乙二胺四乙酸二钠盐,然后升温至190-200℃进行反应,通过气相色谱对反应进程进行监控,当反应釜内物料中2-氯-6-氟三氯苄含量≤5.0%(溶剂除外)后停止反应,得反应后物料ⅰ;
7、3)将步骤2)所得的反应后物料ⅰ通过减压蒸馏,得到石蜡油、2-氯-6-氟一氯苄、2-氯-6-氟二氯苄和2-氯-6-氟三氯苄的混合物,釜底残液作为危废进行处置;减压蒸馏的温度不超过180℃。
8、4)将步骤3)蒸馏所得石蜡油、2-氯-6-氟一氯苄、2-氯-6-氟二氯苄和2-氯-6-氟三氯苄的混合物转入精馏釜进行减压精馏,收集塔顶馏份,分别得到2-氯-6-氟一氯苄和2-氯-6-氟二氯苄,釜底残液为2-氯-6-氟三氯苄和石蜡油的混合物。
9、作为本发明的一种优选方案,步骤2)中,所述的溶剂为石蜡油。
10、作为本发明的一种优选方案,步骤2)中,所述的催化剂为三氯化铁。
11、作为本发明的一种优选方案,步骤2)中,所述溶剂与所述2-氯-6-氟三氯苄的重量比为0.5-1.0:1。
12、作为本发明的一种优选方案,步骤2)中,所述2-氯-6-氟三氯苄与所述乙二胺四乙酸二钠盐的重量比为8-12:1。
13、作为本发明的一种优选方案,步骤3)中,减压蒸馏的真空度为-0.090mpa至-0.01mpa,收集2-氯-6-氟一氯苄时,釜温145-146℃,顶温为120-130℃;收集2-氯-6-氟二氯苄时,釜温146-150℃,顶温为130-134℃。
14、作为本发明的一种优选方案,步骤4)精馏得到2-氯-6-氟一氯苄用于生产农药氟节胺时,其中的2-氯-6-氟一氯苄含量大于99.5%,2-氯-6-氟二氯苄含量小于0.2%。
15、作为本发明的一种优选方案,步骤4)精馏得到2-氯-6-氟二氯苄通过催化水解用于生产2-氯-6-氟苯甲醛时,其中的2-氯-6-氟一氯苄含量小于0.1%、2-氯-6-氟二氯苄含量大于99%,2-氯-6-氟三氯苄小于1%。
16、作为本发明的一种优选方案,步骤4)精馏所得釜底残液混合物返回步骤2)用于代替溶剂。
17、与现有技术相比,本发明具有以下有益效果:
18、本发明的2-氯-6-氟三氯苄脱氯还原方法具有工艺简单、产品收率高和质量优等特点,既解决副产物出路的难题,而且还可以显著降低2-氯-6-氟一氯苄或2-氯-6-氟二氯苄的生产成本,具有很好的经济和社会效益。因此,本发明给2-氯-6-氟二氯苄的合成增加了一个降低原料成本和更加简洁的合成路线。本发明所得的2-氯-6-氟二氯苄后续按照常规方式可用于2-氯-6-氟苯甲醛的合成。
技术特征:
1.一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,以2-氯-6-氟三氯苄为起始原料,在溶剂和催化剂作用下跟乙二胺四乙酸二钠盐进行脱氯还原反应,得到反应混合物;反应所得混合物通过蒸馏和精馏,分离得到产品2-氯-6-氟二氯苄和2-氯-6-氟一氯苄。
2.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,包括以下步骤:
3.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤2)中,所述的溶剂为石蜡油,催化剂为三氯化铁。
4.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤2)中,所述溶剂与所述2-氯-6-氟三氯苄的重量比为0.5-1.0:1。
5.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤2)中,所述2-氯-6-氟三氯苄与所述乙二胺四乙酸二钠盐的重量比为8-12:1。
6.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤3)中,减压蒸馏的真空度为-0.090mpa至-0.01mpa,收集2-氯-6-氟一氯苄时,釜温145-146℃,顶温为120-130℃;收集2-氯-6-氟二氯苄时,釜温146-150℃,顶温为130-134℃。
7.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤4)精馏得到2-氯-6-氟一氯苄用于生产农药氟节胺时,其中的2-氯-6-氟一氯苄含量大于99.5%,2-氯-6-氟二氯苄含量小于0.2%。
8.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤4)精馏得到2-氯-6-氟二氯苄通过催化水解用于生产2-氯-6-氟苯甲醛时,其中的2-氯-6-氟一氯苄含量小于0.1%、2-氯-6-氟二氯苄含量大于99%,2-氯-6-氟三氯苄小于1%。
9.根据权利要求1所述的一种2-氯-6-氟三氯苄脱氯还原方法,其特征在于,步骤4)精馏所得釜底残液混合物返回步骤2)用于代替溶剂和部分原料2-氯6-氟三氯苄。
技术总结
本发明提供了一种2‑氯‑6‑氟三氯苄脱氯还原方法,以2‑氯‑6‑氟三氯苄为起始原料,在溶剂和催化剂作用下跟乙二胺四乙酸二钠盐进行脱氯还原反应,得到反应混合物;反应所得混合物通过蒸馏和精馏,分离得到产品2‑氯‑6‑氟二氯苄和2‑氯‑6‑氟一氯苄。本发明的2‑氯‑6‑氟三氯苄脱氯还原方法具有工艺简单、产品收率高和质量优等特点,既解决副产物出路的难题,还可以显著降低2‑氯‑6‑氟一氯苄或2‑氯‑6‑氟二氯苄的生产成本,具有很好的经济和社会效益。本发明给2‑氯‑6‑氟二氯苄的合成增加了一个降低原料成本和更加简洁的合成路线,所得的2‑氯‑6‑氟二氯苄后续按照常规方式可用于2‑氯‑6‑氟苯甲醛的合成。
技术研发人员:王国平,华慧梁,柯文健,张少美
受保护的技术使用者:浙江大洋生物科技集团股份有限公司
技术研发日:
技术公布日:2024/1/12
- 还没有人留言评论。精彩留言会获得点赞!