含噻唑结构的吲哚啉类化合物及其制备方法和应用
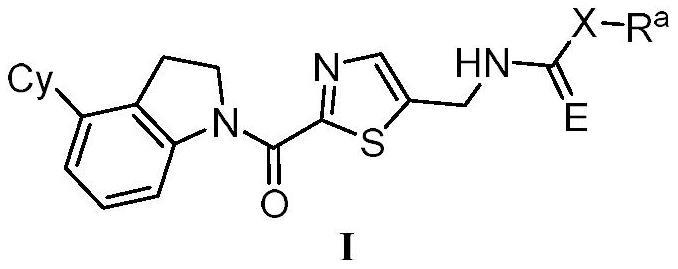
本发明属于医药,涉及通式i所示的含噻唑结构的吲哚啉类化合物或其立体异构体或其药学上可接受的盐,以及它们的制备方法和所述化合物或含有所述化合物的药物组合物在制备治疗与pd-1/pd-l1蛋白/蛋白相互作用、nampt有关的疾病的药物中的应用。
背景技术:
1、近年来,随着对免疫调节分子机制的阐明,肿瘤免疫治疗迅速发展并取得良好的临床疗效。程序性细胞死亡受体1(programmed cell death-1,pd-1)是t细胞表面受体,当其与程序性细胞死亡配体1(programmed cell death-ligand 1,pd-l1)结合时,产生负性免疫调节信号,从而抑制t细胞活化、增殖以及白细胞介素2(il-2)和干扰素γ(ifn-γ)等细胞因子的分泌。大量研究表明,机体内的肿瘤微环境会诱导浸润的t细胞中pd-1表达上调,同时肿瘤细胞高表达pd-l1,导致pd-1/pd-l1介导的信号通路持续激活,肿瘤特异性cd8+t细胞功能被抑制,以至于无法识别或杀伤肿瘤细胞,即造成肿瘤细胞免疫逃逸。因此靶向阻断pd-1/pd-l1蛋白/蛋白相互作用,可以有效恢复t细胞功能,使其重新识别并杀伤肿瘤细胞。
2、基于pd-1/pd-ll的免疫疗法备受瞩目,目前已被批准上市的单抗药物包括默沙东的pembrolizumab、百时美施贵宝的nivolumab、默克的avelumab、阿斯利康的durvalumab、罗氏的atezolizumab、再生元/赛诺菲的cemiplimab和葛兰素史克的dostarlimab。上述药物已在多种肿瘤治疗中显示出明显疗效,被批准的适应症包括黑色素瘤、非小细胞肺癌、胃癌、尿路上皮癌等。随着临床研究的开展,单抗药物有望在更多的适应症中实现突破。
3、虽然单抗药物在临床治疗中显示出优势,但也存在明显的缺陷,如制备和纯化困难、生产成本高昂、易被蛋白酶分解、无法口服给药以及与单抗免疫原性相关的严重毒副作用。相比于生物大分子药物,小分子化合物经化学修饰后药物代谢动力学性质可控,同时在生产工艺、给药方式等方面也具有更大的探索与优化空间。同时,接受pd-1/pd-l1单抗治疗的患者存在广泛的原发性或继发性耐药,针对耐药进行创新药物研究有望改善肿瘤免疫治疗中的瓶颈问题。耐药的发生与多种因素相关,目前已确证肿瘤细胞高代谢导致t细胞增殖受到抑制,并促进肿瘤免疫微环境的形成,极大降低了单抗药物的疗效。因此,适当地干预肿瘤细胞代谢、改善肿瘤免疫微环境可以增强抗肿瘤免疫应答,提高抗pd-1/pd-l1治疗临床疗效。
4、烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,nad+)是细胞氧化还原反应中的重要辅酶之一,在多种细胞生理过程尤其是能量代谢过程中发挥着至关重要的作用。除此之外,nad+是去乙酰化酶sirtuins、多聚adp核糖聚合酶1和adp环化酶的底物,对ca+动员、基因稳定、细胞凋亡、代谢等方面有重要影响。为了维持稳定的细胞内nad+浓度,生物主要通过以下几个途径合成nad+:①由色氨酸从头合成。②由烟酸和烟酰胺核糖体旁合成。③经回收nam(nad+代谢产物)进而补救回收合成nmn。其中,补救回收合成是nad+生物合成的一个重要途径,而烟酰胺磷酸核糖转移酶(nicotinamidephosphoribosyltransferase,nampt)是该途径的限速酶。
5、肿瘤细胞的新陈代谢非常活跃,比正常细胞消耗nad+的速度更快,因此在肿瘤代谢中,肿瘤细胞会通过高表达nampt以满足对nad+的高需求。因此,在一些nampt过表达的肿瘤中,通过有效抑制nampt会直接抑制肿瘤细胞内nad+的产生,进而导致细胞死亡。另一方面,正常细胞不受nampt抑制的影响,因为它们可以通过其他生物合成途径,如经烟酸磷酸核糖转移酶(nicotinic acid phosphoribosyltransferase,naprt)催化,由烟酸合成nad+。然而,许多癌细胞系由于缺乏naprt,无法通过该途径合成nad+。因此,抑制nampt已成为一种有效、选择性抑制癌细胞生长的策略,代表性nampt抑制剂有chs-828和fk866。
6、
7、研究表明,肿瘤细胞中过表达的nampt会促使免疫抑制性细胞mdscs的扩增,促进免疫抑制微环境的形成。同时,肿瘤细胞糖酵解代谢产物乳酸的堆积可以促使单核细胞分化为树突状细胞,通过分泌免疫抑制因子、抑制t细胞免疫应答而直接抑制免疫反应。因此,肿瘤细胞高代谢导致免疫抑制微环境的形成是抗pd-1/pd-l1治疗获得性耐药发生的重要原因之一。研究表明,与单独治疗相比,联合使用pd-1单抗药物和nampt抑制剂,肿瘤体积和重量明显降低,显示出更好的抗肿瘤活性(travelli c et al.cancer research,2019,79(8):1938-1951)。同时抑制pd-1/pd-l1相互作用和nampt从理论上具有协同的免疫活化功能,并有望在一定程度上克服pd-1/pd-l1抑制剂耐药和nampt抑制剂剂量限制性毒性问题。因此,针对性地开发pd-1/pd-l1相互作用和nampt双重抑制剂将有望获得抗肿瘤活性突出、作用机制独特的化合物,具有重要的研究意义。
技术实现思路
1、本发明的首要目的是提供通式i所示的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐,
2、
3、其中,
4、cy选自苯基、所述苯基可任选被1–3个r1取代;
5、r1独立地选自氢、卤素、氰基、羟基、羧基、氨基、(c1–c4)烷基、(c1–c4)烷氧基、(c1–c4)烷基甲酰基氨基;所述的(c1–c4)烷基、(c1–c4)烷氧基、(c1–c4)烷基甲酰基氨基可任选被1–3个取代;
6、r2、r3独立地选自氢、(c1–c4)烷基、羟基(c1–c4)烷基;
7、或者r2、r3和与它们相连的氮原子一起形成一个3–6元的含氮杂环,优选为4–6元含氮杂环;所述的含氮杂环含有1–3个选自n、o和s的杂原子;所述的含氮杂环可任选被1–3个r4取代;
8、r4独立地选自氢、羟基、羧基、(c1–c4)烷基、羟基(c1–c4)烷基;
9、e选自o、n–cn;
10、x选自氨基(c1–c4)烷基、(c2–c4)烯基、
11、ra选自苯基、吡啶基;所述的苯基、吡啶基可任选被1–3个r5取代;
12、r5独立地选自氢、卤素、羟基、氨基、(c1–c4)烷基;
13、本发明优选涉及通式i的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐,
14、其中,
15、cy选自苯基、所述苯基可任选被1–3个r1取代;
16、r1独立地选自氢、卤素、羟基、氨基、(c1–c4)烷基、(c1–c4)烷氧基、(c1–c4)烷基甲酰基氨基;所述的(c1–c4)烷氧基、(c1–c4)烷基甲酰基氨基可任选被1–3个取代;
17、选自:
18、
19、
20、e选自o、n–cn;
21、x选自氨基(c1–c4)烷基、(c2–c4)烯基、
22、ra独立地选自苯基、吡啶基;所述的苯基、吡啶基可任选被1–3个氢或卤素取代。
23、本发明进一步优选涉及通式i所示的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐,
24、其中,
25、cy选自苯基、所述苯基可任选被1–3个r1取代;
26、r1独立地选自氢、卤素、羟基、氨基、甲基、甲氧基、乙氧基、丙氧基、丙酰基氨基;所述的乙氧基、丙氧基、丙酰基氨基可任选被1–3个取代;
27、选自:
28、
29、当e为o时,x选自当e为n–cn时,x为
30、ra为吡啶基;所述吡啶基可任选被1–3个氢或卤素取代。
31、本发明通式i所示的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐最终优选自以下化合物:
32、
33、
34、
35、此外,本发明还包括本发明所述化合物的前药。本发明所述化合物的前药是通式i所示化合物的衍生物,它们自身可能具有较弱的活性甚至没有活性,但是在给药后,在生理条件下(例如通过代谢、溶剂分解或另外的方式)被转化成相应的生物活性形式。
36、以上所述的通式i的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐中,所述的药学上可接受的盐包括与无机酸、有机酸、碱金属离子形成的盐;所述的无机酸选自:盐酸、氢溴酸、氢氟酸、硫酸、磷酸;所述的有机酸选自:琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、甲磺酸、乙磺酸、对甲苯磺酸;所属的碱金属离子选自锂离子、钠离子、钾离子。
37、本发明中“卤素”是指氟、氯、溴或碘;“烷基”是指直链或支链的烷基;代表取代基连接处。
38、本发明通式i所示的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐作为活性成分,与药学上可接受的载体或赋形剂混合制备成药物组合物,所述载体或赋形剂包括本领域公知的稀释剂、粘合剂、润湿剂、崩解剂、润滑剂、助流剂。稀释剂包括淀粉、糊精、蔗糖、葡萄糖、乳糖、甘露醇、山梨醇、木糖醇、磷酸氢钙;湿润剂包括水、乙醇、异丙醇;粘合剂包括淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、羟丙基甲基纤维素、乙基纤维素、聚乙二醇;崩解剂包括干淀粉、微品纤维素、低取代羟丙基纤维素、交联聚乙烯吡咯烷酮、交联羧甲基纤维素钠、羧甲基淀粉钠、十二烷基磺酸钠;润滑剂和助流剂包括滑石粉、二氧化硅、聚乙二醇。
39、本发明的药物组合物可配制成若干种剂型,所述剂型选自注射剂、片剂、胶囊剂。
40、本发明所述的含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐可以与其他活性成分组合使用,从而达到更优的治疗效果。
41、本发明还提供了所述含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐在制备预防和/或治疗与pd-1/pd-l1蛋白/蛋白相互作用和nampt有关的疾病的药物中的应用。所述的pd-1/pd-l1蛋白/蛋白相互作用和nampt有关的疾病选自癌症、感染性疾病。所述的癌症选自淋巴瘤、非小细胞肺癌、小细胞肺癌、头颈部细胞癌、神经胶质瘤、成神经细胞瘤、肺鳞癌、肺腺癌、膀胱癌、胃癌、结肠癌、大肠癌、肾癌、胆管癌、胃癌、食管鳞癌、卵巢癌、胰腺癌、乳腺癌、前列腺癌、肝癌、脑癌、黑色素瘤、多发性骨髓瘤、皮肤癌、上皮细胞癌、白血病和宫颈癌;所述的感染性疾病选自细菌感染、病毒感染。
42、本发明还提供所述通式i所示化合物的制备方法。所有的原料及中间体都是通过如下流程中描述的方式,通过有机化学领域普通技术人员熟知的方法制备的或者商购。
43、路线一:
44、
45、(a)以4-溴-1h-吲哚为起始原料,在还原剂如氰基硼氢化钠作用下制得中间体2;
46、(b)以中间体2和5-(1,3-二氧戊环-2-基)噻唑-2-羧酸为原料,在缩合剂条件下经过酰胺化反应制得中间体3;
47、(c)以中间体3为原料,在对甲苯磺酸的作用下脱保护制得含有甲酰基的中间体4;
48、(d)以中间体4为原料,在还原剂如硼氢化钠作用下制得中间体5;
49、(e)以中间体5和苯、取代苯环硼酸或硼酸酯为原料,通过suzuki-miyaura偶联反应制得中间体6;
50、(f)以中间体6为原料,在氯代试剂如二氯亚砜的作用下制得中间体7;
51、(g)以中间体7为原料,通过gabriel反应制得中间体8;
52、(h)以中间体8为原料,通过亲核取代或酰胺化反应制得通式i的目标化合物。
53、路线二:
54、
55、(i)以中间体5和间羟基苯硼酸为原料,通过suzuki-miyaura偶联反应制得中间体9;
56、(j)以中间体9为原料,与二卤代物经取代反应制得中间体10;
57、(k)以中间体10为原料,与小分子胺经取代反应制得中间体11;
58、(l)以中间体11为原料,在氯代试剂如二氯亚砜的作用下制得中间体12;
59、(m)以中间体12为原料,通过gabriel反应制得中间体13;
60、(n)以中间体13为原料,通过亲核取代或酰胺化反应制得通式i的目标化合物。
61、路线三:
62、
63、(o)以中间体9为原料,在氯代试剂如二氯亚砜的作用下制得中间体14;
64、(p)以中间体14为原料,通过gabriel反应制得中间体15;
65、(q)以中间体15为原料,通过亲核取代或酰胺化反应制得中间体16。
66、(r)以中间体16为原料,与二卤代物经取代反应制得中间体17;
67、(s)以中间体17为原料,与小分子胺经取代反应制得通式i的目标化合物。
68、路线四:
69、
70、(t)以中间体5和间氨基苯硼酸为原料,通过suzuki-miyaura偶联反应制得中间体18;
71、(u)以中间体18为原料,在氯代试剂如二氯亚砜的作用下制得中间体19;
72、(v)以中间体19为原料,通过gabriel反应制得中间体20;
73、(w)以中间体20为原料,通过亲核取代或酰胺化反应制得中间体21;
74、(x)以中间体21为原料,与卤代烷基酰卤反应制得中间体22;
75、(y)以中间体22为原料,与小分子胺经取代反应制得通式i的目标化合物。
76、所述的cy、e、x、ra的定义如权利要求所述;所述n为1~3,y为氯或溴。本发明的具有通式i的含噻唑结构的吲哚啉类化合物均可按照上述反应路线描述的方法或类似的方法制备得到。
77、本发明的有益效果:
78、本发明提供的通式i所示的化合物对pd-1/pd-l1蛋白/蛋白相互作用和nampt具有高水平的抑制活性,对nampt高表达的a2780细胞具有显著的抗增殖活性,代表性化合物能在过表达nampt、pd-l1的动物模型中抑制肿瘤生长。因此,本发明所述含噻唑结构的吲哚啉类化合物、其立体异构体或其药学上可接受的盐,或含有所述化合物的药物组合物能够用于制备治疗与pd-1/pd-l1蛋白/蛋白相互作用、nampt有关的疾病如癌症、病毒感染的药物。
- 还没有人留言评论。精彩留言会获得点赞!