一种4-亚苄基吡唑酮衍生物及其合成方法
本发明涉及一种4-亚苄基吡唑酮衍生物及其合成方法,属于有机合成。
背景技术:
1、吡唑酮结构在天然产物及药物分子中普遍存在,探索这类化合物的高效合成方法一直是有机合成研究人员的研究兴趣所在。4-亚苄基吡唑酮衍生物代表了一类重要的吡唑酮化合物,这类化合物具有丰富的药物活性,如抗癌活性(chen,l.etal.j.med.chem.2012,55,7037),hiv-1整合酶抑制剂(neamati,n.etal.j.med.chem.2008,51,1136),以及h1n1和h5n1神经氨酸酶抑制剂(liang,p.-h.org.lett.2014,16,5060);不仅如此,这类化合物还是合成先导化合物和功能分子的重要前体(yang,l.and zhong,g.et al.org.lett.2019,21,7943;feng,x.etal.org.lett.2019,21,1632;li,e.-q.and duan,z.et al.adv.synth.catal.2019,361,1389);此外,这类化合物也被广泛用作配体(li,f.et al.j.am.chem.soc.2011,133,15276)或染料(cui,y.-p.et al.dyespigm.,2009,81,27)。
2、鉴于此,发展简洁、高效的合成4-亚苄基吡唑酮衍生物的方法,具有较高的应用价值。传统的合成方法依赖于吡唑酮和芳香醛、酮发生knoevenagel反应(cristea,i.etal.heterocyl.commun.1998,4,139;shingare,m.s.et al.synth.commun.2002,32,497),通常需要回流或微波加热条件,尽管使用离子液体作为溶剂可以降低反应温度至室温,但这一条件仅适用于芳香醛类底物(shingare,m.s.et al.greenchem.2002,4,266),具有较大的局限性;n-丙炔酰腙类化合物在金催化剂存在下也可以经由分子内环异构化/1,3-迁移的串联过程转化为4-亚苄基吡唑酮衍生物(zhan,z.-p.et al.j.org.chem.2015,80,9307),为4-亚苄基吡唑酮衍生物的合成提供了新的途径,但需要使用过渡金属催化剂和底物合成路线繁琐等缺点限制了该方法的应用,此外,该反应同样经历了knoevenagel缩合过程,反应温度较高(>100℃);在亚当量碘存在下,吡唑酮可以快速与反式2-羟基查尔酮发生串联的michael加成/环异构化/氧化脱氢反应,生成4-亚苄基吡唑酮产物(bakthadoss,m.et al.tetrahedron2018,74,490),尽管该反应效率较高,但同样需要高温条件(>100℃);近期,三乙烯二胺(dabco)催化morita–baylis–hillman(mbh)碳酸酯与2-亚烯丙基丙二腈的[5+1]环化反应为4-亚苄基吡唑酮衍生物的合成提供了一条温和的路径(han,b.andzhan,g.et al.new j.chem.2022,46,11617),然而,复杂的底物合成路线和产物固定的取代模式降低了该反应的实用性;1,3-二芳基丙烯与吡唑酮的氧化偶联反应同样可以在室温下构筑4-亚苄基吡唑酮结构(li,j.,xu,x.and cheng,d.et al.eur.j.org.chem.2023,26,e202201480),然而,该方法仅适用于合成亚苄位烯基取代的4-亚苄基吡唑酮衍生物。
3、因此,发展温和条件下,从简单、稳定、易得的原料出发,合成亚苄位具有不同取代模式的4-亚苄基吡唑酮衍生物的方法,仍然是有机合成中一个值得研究并富有挑战性的课题。
技术实现思路
1、本发明旨在提供一种4-亚苄基吡唑酮衍生物及其合成方法。
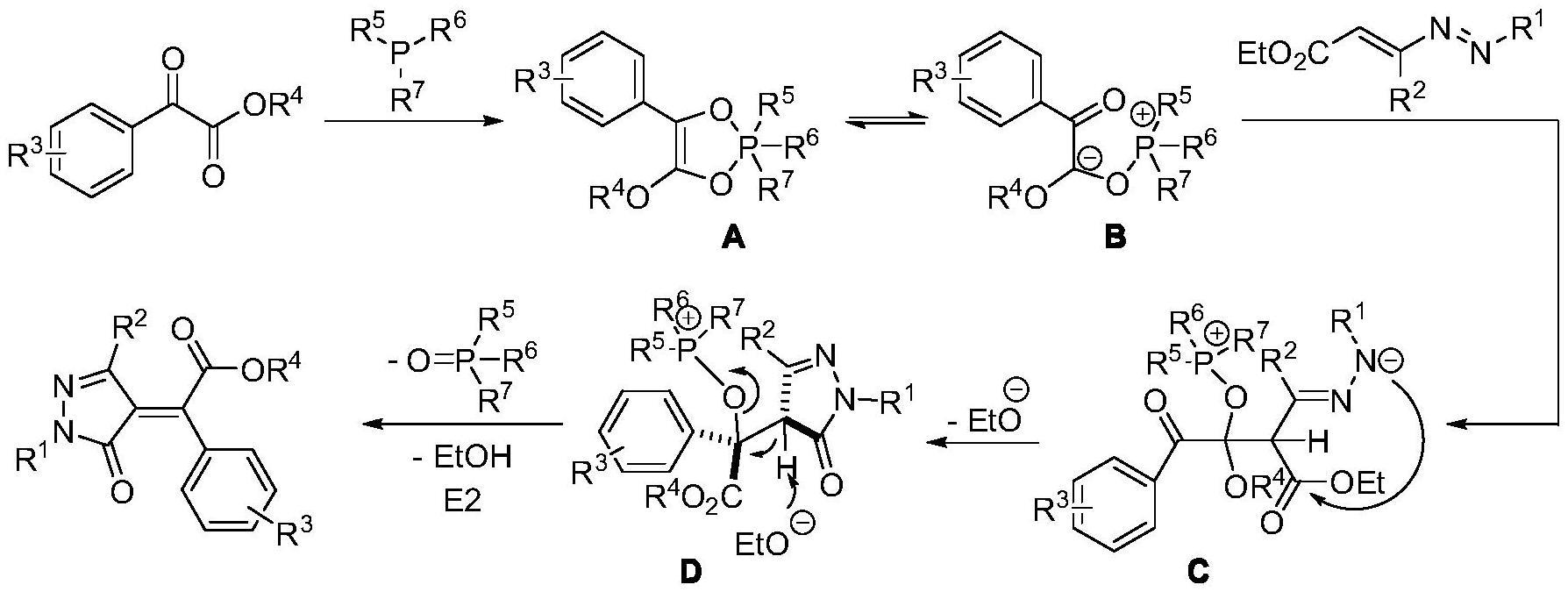
2、本发明的机理为:磷试剂与α-酮酸酯反应,形成kukhtin-ramirez加合物a;作为a的共振式,偶极体b随后亲核进攻1,2-二氮-1,3-丁二烯化合物,形成中间体c;c经由分子内酯交换过程转化为中间体d,并在乙氧基负离子的作用下发生e2消除,脱去氧化膦的同时生成4-亚苄基吡唑酮产物。具体过程如下式所示:
3、
4、本发明提供了一种4-亚苄基吡唑酮衍生物,具有如式i所示的结构式:
5、i
6、
7、4-亚苄基吡唑酮衍生物是通过1,2-二氮-1,3-丁二烯化合物ii
8、ii
9、
10、与α-酮酸酯iii
11、iii
12、
13、在磷试剂iv
14、iv
15、的促进下反应得到。
16、其反应通式为:
17、
18、上述结构式中r1选自芳基,r3选自氢、卤素、烷基或烷氧基;r2和r4选自烷基或芳基;r5、r6和r7选自二(烷基)氨基、烷基或芳基。
19、本发明提供了一种4-亚苄基吡唑酮衍生物的合成方法,包括以下合成步骤:
20、将α-酮酸酯及1,2-二氮-1,3-丁二烯化合物溶于有机溶剂中,所得反应混合物置于低温下搅拌10~15分钟,随后将稀释的磷试剂在5~15分钟内滴加至上述反应混合物中,α-酮酸酯、1,2-二氮-1,3-丁二烯化合物及磷试剂的摩尔投料比为1:1~2:1~1.2,滴加完毕后将反应自然升温至室温,继续搅拌12小时,反应完成后旋蒸除去溶剂,粗产品经200~300目硅胶柱层析纯化,采用石油醚-乙酸乙酯体积比为30:1~15:1的混合溶液作洗脱液,所得纯品计算收率,根据目标化合物不同,收率为38~69%。
21、上述合成方法中,所述有机溶剂包括非极性溶剂甲苯;极性溶剂四氢呋喃、二氯甲烷、氯仿、乙腈、乙酸乙酯中的一种,有机溶剂的用量为5-20ml/mmol的α-酮酸酯。
22、上述合成方法中,所述低温采用-45~-78℃。
23、上述合成方法中,所述磷试剂包括六甲基亚磷酰三胺、三正丁基膦、三环己基膦、三苯基膦以及包含磷氮键和磷碳键的其他三配位磷试剂中的一种。
24、上述合成方法中,磷试剂用有机溶剂稀释后加入反应体系中,所述有机溶剂与反应体系中的有机溶剂相同,包括甲苯、四氢呋喃、二氯甲烷、氯仿、乙腈、乙酸乙酯中的一种;稀释的磷试剂浓度为0.20~0.24mol/l。
25、本发明的有益效果:
26、(1)本发明所用原料简单易得,稳定性好;
27、(2)反应条件温和,且4-亚苄基吡唑酮产物上取代基的种类丰富,可以灵活变换;
28、(3)本发明所合成的化合物具有丰富的药物活性,为含4-亚苄基吡唑酮结构的新型药物分子的设计合成和开发提供了候选化合物。
技术特征:
1.一种4-亚苄基吡唑酮衍生物,其特征在于:是通过1,2-二氮-1,3-丁二烯化合物与α-酮酸酯在磷试剂的促进下反应得到;所得4-亚苄基吡唑酮衍生物具有如i所示的结构式:
2.根据权利要求1所述的4-亚苄基吡唑酮衍生物,其特征在于:所述1,2-二氮-1,3-丁二烯化合物具有下式ii所示的结构式:
3.一种权利要求1或2所述的4-亚苄基吡唑酮衍生物的合成方法,其特征在于合成步骤如下:将α-酮酸酯及1,2-二氮-1,3-丁二烯化合物溶于有机溶剂中,所得反应混合物置于低温下搅拌10~15分钟,随后将稀释的磷试剂在5~15分钟内滴加至上述反应混合物中,α-酮酸酯、1,2-二氮-1,3-丁二烯化合物及磷试剂的摩尔投料比为1∶1~2∶1~1.2,滴加完毕后将反应体系自然升温至室温,继续搅拌12小时,反应完成后旋蒸除去溶剂,粗产品经200~300目硅胶柱层析纯化,采用石油醚-乙酸乙酯体积比为30:1~15:1的混合溶液作洗脱液,得到4-亚苄基吡唑酮衍生物。
4.根据权利要求3所述的4-亚苄基吡唑酮衍生物的合成方法,其特征在于:所述有机溶剂包括甲苯、四氢呋喃、二氯甲烷、氯仿、乙腈、乙酸乙酯中的任一种,有机溶剂的用量为5-20 ml/mmol的α-酮酸酯。
5.根据权利要求3所述的4-亚苄基吡唑酮衍生物的合成方法,其特征在于:所述低温采用-45~-78 oc。
6.根据权利要求3所述的4-亚苄基吡唑酮衍生物的合成方法,其特征在于:所述磷试剂包括六甲基亚磷酰三胺、三正丁基膦、三环己基膦、三苯基膦中的任一种。
7.根据权利要求3所述的4-亚苄基吡唑酮衍生物的合成方法,其特征在于:磷试剂用有机溶剂稀释后加入反应体系中,所述有机溶剂与反应体系中的有机溶剂相同,稀释的磷试剂浓度为0.20~0.24 mol/l。
技术总结
本发明公开了一种4‑亚苄基吡唑酮衍生物及其合成方法,属于有机合成技术领域。通过a‑酮酸酯与1,2‑二氮‑1,3‑丁二烯化合物在磷试剂的促进下反应得到4‑亚苄基吡唑酮衍生物;本发明所用原料简单易得,稳定性好;反应条件温和,且合成的4‑亚苄基吡唑酮产物上取代基的种类丰富,可以灵活变换;本发明提供的4‑亚苄基吡唑酮衍生物具有丰富的药物活性,为含4‑亚苄基吡唑酮结构的新型药物分子的设计合成和开发提供了候选化合物。
技术研发人员:周荣,郭宏宇,杜云凤
受保护的技术使用者:太原理工大学
技术研发日:
技术公布日:2024/1/14
- 还没有人留言评论。精彩留言会获得点赞!