一种基于EGFR变构位点的PROTAC类化合物及应用
本发明涉及药物化合物合成领域,具体涉及一种基于egfr变构位点的protac类化合物及应用。
背景技术:
1、蛋白降解靶向嵌合体(proteolysis targeting chimeras,protac)是目前发展较快且相对成熟的一种靶向蛋白降解技术,其降解靶蛋白(protein of interest,poi)是通过ups实现的。protac是一种异双功能分子,一端是poi的配体,另一端是e3连接酶的配体,两个配体通过连接子(linker)相连。当protac进入细胞后,可将poi拴在特定的e3连接酶亚单位上,形成poi-protac-e3三元复合物,并触发k48多泛素化,导致其随后被蛋白酶体降解。理论上,protac只需与致病蛋白短暂结合(非长久抑制),给蛋白打上泛素化标签即可释放出来,因此其作用类似于催化剂可以循环利用,这使得protac在细胞内发挥作用只需很小的浓度。
2、已知的e3连接酶有600余种,但目前应用于protac技术的e3连接酶主要有四种:von hippel-lindau(vhl),cereblon(crbn),murine double mimute2(mdm2)和cellularinhibitors of apoptosis proteins 1(ciap1)。
3、egfr(epidermal growth factor receptor,简称为egfr、erbb-1或her1)是表皮生长因子受体(her)家族成员之一,该家族包括her1(erbb1,egfr)、her2(erbb2,neu)、her3(erbb3)及her4(erbb4),her家族在细胞生理过程中发挥重要的调节作用。egfr广泛分布于哺乳动物上皮细胞、成纤维细胞、胶质细胞、角质细胞等细胞表面,egfr信号通路对细胞的生长、增殖和分化等生理过程发挥重要的作用。
4、研究表明:在许多实体肿瘤中存在egfr的高表达或异常表达,egfr与肿瘤细胞的增殖、血管生成、肿瘤侵袭、转移及细胞凋亡的抑制有关。其可能机制有:egfr的高表达引起下游信号传导的增强;突变型egfr受体或配体表达的增加导致egfr的持续活化;自分泌环的作用增强;受体下调机制的破坏;异常信号传导通路的激活等。egfr的过表达在恶性肿瘤的演进中起重要作用,胶质细胞、肾癌、肺癌、前列腺癌、胰腺癌、乳腺癌等组织中都有egfr的过表达,因此其已成为有效的抗肿瘤靶点。
5、目前,多个egfr酪氨酸激酶抑制剂(tyrosine kinase inhibitors,tkis)已经上市用于多种癌症的治疗,可以分为三代:第一代为单靶点可逆egfr靶向酪氨酸激酶抑制剂,对19外显子缺失突变(egfr19 del/delins)和21外显子858密码子错义突变(egfr21l858r)有效,代表药物有吉非替尼(易瑞沙)、厄洛替尼、埃克替尼;第二代是egfr和其他erbb酪氨酸激酶受体家族成员的不可逆共价抑制剂,对egfr典型的敏感型突变(egfr19del/delins、21l858r)和“中度敏感”突变(g719x、s768i、l861q)以及一些罕见突变有效,代表药物有阿法替尼、达克替尼、拉帕替尼;第三代同时靶向egfr敏感突变和t790m耐药突变,且可有效穿透血脑屏障,对脑转移效果显著,代表药物有奥希替尼和国产的阿美替尼。
6、但是,在接受8-13个月的egfr-tkis治疗后,几乎所有的患者都不可避免地出现获得性耐药,降低了egfr-tkis的抗肿瘤效果。
7、第四代egfr抑制剂eai045结合于egfr的变构位点而非atp结合位点,因此atp结合位点内的氨基酸突变对eai045与egfr的结合影响较小,并且文献也已证eai045对egfr野生型以及常见突变型均有较好的结合能力,但仍未完全解决小分子egfr抑制剂的突变耐药问题。
技术实现思路
1、本发明的目的在于提供了一种基于egfr变构位点的protac类化合物及应用,该化合物不仅具有优异的egfr蛋白降解作用和抗癌活性,还能减轻对人体的毒副作用,可用于制备抗肿瘤药物。
2、本发明的另一目的在于提供了上述化合物或其药学上可接受的盐和水合物在制备预防或/和治疗癌症的药物中的应用。
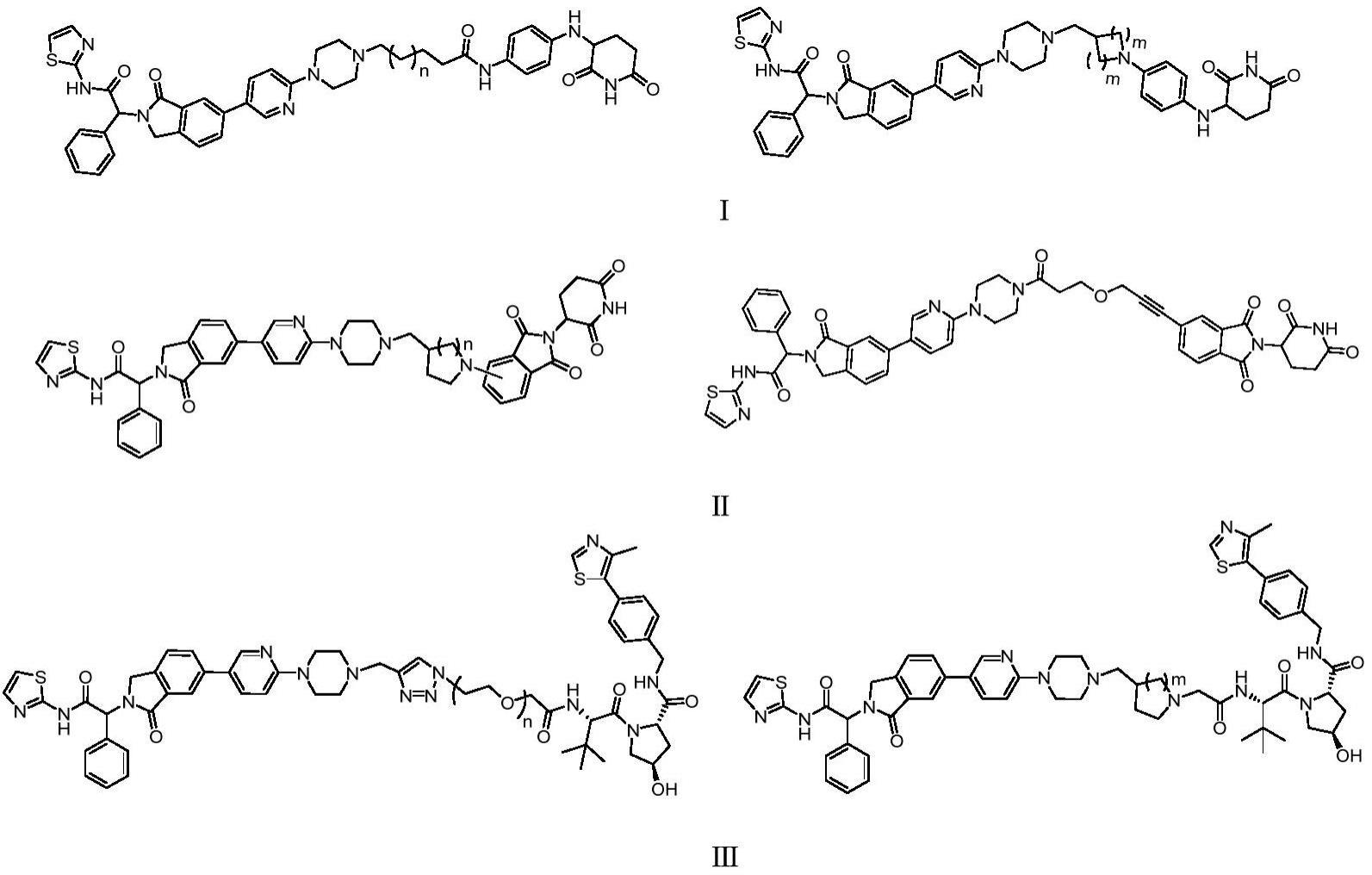
3、为了实现上述发明目的,本发明提供了具有下式i、ⅱ、ⅲ所示的化合物或其药学上可接受的盐和水合物:
4、
5、其中,式ⅰ化合物n为1~7的整数,m为1~2的整数;式ⅱ化合物n为1~2的整数;式ⅲ化合物n为1~7的整数,m为1~2的整数。
6、本发明通过使用连接链将egfr小分子抑制剂和e3泛素连接酶复合体中crbn蛋白配体或vhl蛋白配体进行连接制得一种蛋白降解靶向联合体(protacs)双功能小分子,通过对egfr进行泛素化标记,能够选择性诱导egfr蛋白降解,具有较好的抗肿瘤活性。
7、优选地,式ⅰ化合物为刚性链时活性更好,进一步优选m=1;式ⅱ化合物n为1~2的整数;式ⅲ化合物n为1~7的整数,m为1~2的整数。n为1~7的整数,进一步优选为2~5的整数,优选的化合物具有较好的诱导egfr降解作用和抗肿瘤活性。
8、本发明还包括式(i、ⅱ、ⅲ)所示的化合物的立体异构体。本发明化合物的所有立体异构体,包括但不限于非对映异构体、对映异构体和阻转异构体以及他们的混合物(如外消旋物),均包括在本发明的范围内。
9、本发明还包括式(i、ⅱ、ⅲ)所示的化合物的互变异构体。属于“互变异构体”或“互变异构形式”是指经由低能垒相互转化的不同能量的结构异构体。
10、本发明还包括式(i、ⅱ、ⅲ)的衍生物的前药,式(i、ⅱ、ⅲ)的衍生物自身可能具有较弱的活性甚至没有活性,但是在给药后,在生理条件下(例如通过代谢、溶剂分解或另外的方式)被转化成相应的生物活性形式。
11、药学上可接受的盐包括:与下列酸形成的加成盐:盐酸、氢嗅酸、硫酸、磷酸、甲磺酸、乙磺酸、对甲苯磺酸、苯磺酸、茶二磺酸、乙酸、丙酸、乳酸、三氟乙酸、马来酸、柠檬酸、富马酸、草酸、酒石酸或苯甲酸;以及盐酸、氢嗅酸、硫酸、柠檬酸、酒石酸、磷酸、乳酸、丙酮酸、乙酸、三氟乙酸、马来酸、苯磺酸或琉拍酸的酸成盐。
12、式ⅰ柔性链化合物通过如下反应式制得:
13、
14、式ⅰ刚性链化合物通过如下反应式制得:
15、
16、式ⅱ化合物通过如下反应式制得:
17、
18、
19、式ⅲ柔性链化合物通过如下反应式制得:
20、
21、式ⅲ刚性链化合物通过如下反应式制得:
22、
23、一种药物组合物,包括式(i、ⅱ、ⅲ)所述的化合物或其药学上可接受的盐和水合物,和药学上可接受的赋形剂。
24、其中,式(i、ⅱ、ⅲ)所述的化合物或其药学上可接受的盐和水合物作为活性成份,与药学上可接受的赋形剂混合制成药物组合物,所述的赋形剂为用于药学领域的稀释剂、辅助剂或载体。
25、一种在药物组合物中加入药学上可接受的辅料制成的临床上可接受的剂型,所述的剂型为注射剂、片剂或胶囊剂。
26、一种药物组合物,包括式(i、ⅱ、ⅲ)所述的化合物或其药学上可接受的盐和水合物,和不同的抗肿瘤药剂。本发明所述的化合物或其药学上可接受的盐、水合物和前药可作为抗肿瘤药剂单独使用,还可以与不同的抗肿瘤药剂联合使用,用于治疗预防肿瘤。
27、本发明还公开了式(i、ⅱ、ⅲ)所示的化合物或其药学上可接受的盐和水合物在制备预防或/和治疗癌症的药物中的应用。
28、所述的癌症为多发性骨髓瘤、胃癌、肺癌、乳腺癌、食管癌、结肠癌、髓母细胞瘤、急性粒细胞白血病、慢性白血病、前列腺癌、肝细胞瘤、肾细胞瘤、宫颈癌、皮肤癌、卵巢癌、结肠癌、神经胶质瘤、甲状腺癌或胰腺癌。
29、本发明的有益效果:
30、本发明放弃“占据驱动(occupancy-driven)”atp结合位点抑制egfr的小分子抑制剂方式,而通过暴露于egfr二聚体外侧的变构位点,由protac触发“事件驱动(event-driven)”降解egfr,解决因egfr的atp结合位点氨基酸残基突变引起的耐药问题。
31、本发明式(i、ⅱ、ⅲ)所述的双功能小分子可以对egfr进行泛素化标记,仅需较少用量即可诱导蛋白降解,这个过程类似于催化反应,并不需要等摩尔量的药物,能减轻对人体的毒副作用;
32、本发明体外抗肿瘤活性测试及体外egfr蛋白降解活性测试表明,式(i、ⅱ、ⅲ)所述的双功能小分子表现出了优异的egfr蛋白降解作用和抗癌活性,其抗癌效果优于egfr抑制剂,可用于制备预防或/和治疗多种癌症的药物,在医药领域具有巨大的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!