一种相变微胶囊及其制备方法和应用与流程
本发明涉及一种相变微胶囊及其制备方法和应用,属于高分子材料领域。
背景技术:
皮革是由动物的原皮加工而成。皮革的调温性能是影响皮革制品舒适性的重要因素。皮革是天然的多孔型的载体,具有较强的吸附性能和吸附容量,非常适合引入相变材料(phasechangematerials,pcm)来改善其调温性能。
相变材料在相变过程中能吸收和释放相变潜热,可广泛应用于能量贮存和温度控制领域,但很多相变材料在应用的过程中存在着相变材料的泄露、相分离以及腐蚀性等问题,解决这些问题的有效办法之一就是在相变材料表面包覆一层性能稳定的高分子膜而构成具有核壳结构的复合材料。现有技术中,可以采用二氧化硅凝胶包覆十二醇制备相变微胶囊,并将其应用到皮革中,具有一定的调温效果。但是,制得的相变微胶囊粒径较大,包封率较差,导热性能也不理想。
中国专利cn105056854a公开了一种纳米tio2改性复合相变微胶囊的制备方法,用正烷烃、硬脂酸烷基酯制备芯材,用甲基丙烯酸,用甲基丙烯酸甲酯、丙烯酸丁酯、丙烯酸乙酯、交联剂和纳米tio2制备出壁材,将芯材与壁材混合得到油相a;将反应性乳化剂溶解于去离子水中制备水相b;将油相a加入水相b中,经超声分散处理形成o/w型预乳液进行加热,并向o/w型预乳液中添加引发剂,制备出纳米tio2改性复合相变微胶囊乳液;将纳米tio2改性复合相变微胶囊。其制备方法复杂,且芯材中含有正烷烃和硬脂酸烷基酯,乳化效果较差。另外,其制得的纳米tio2改性复合相变微胶囊的相变潜热较低,仅为73.5j/g。
技术实现要素:
发明要解决的问题
本发明的目的在于提供一种相变微胶囊及其制备方法和应用。该相变微胶囊的包封率高和导热性能优异,粒径小。进一步地,相变微胶囊应用在皮革中可以更好的渗透和使用。
用于解决问题的方案
本发明提供一种相变微胶囊的制备方法,包括以下步骤:
水相制备步骤:将乳化剂、任选存在的助乳化剂以及任选存在的调节剂溶于水中,分散后得到水相;
油相制备步骤:将单体、芯材、引发剂以及纳米添加剂混合均匀,得到油相;
预乳化步骤:将油相与水相混合,常温下分散并进行预乳化,得到预乳化液;
乳化步骤:将预乳化液进行超声分散,然后在氮气保护下,在75-85℃下加热,反应7-8h后,得到乳化液;
破乳步骤:在乳化液中加入破乳剂进行破乳。
根据本发明的相变微胶囊的制备方法,所述水相制备步骤中,所述乳化剂包括十二烷基硫酸钠和/或烯丙氧基壬基酚聚氧乙烯(10)醚硫酸铵。
根据本发明的相变微胶囊的制备方法,所述水相制备步骤中,所述助乳化剂包括十六烷、十六醇和十八醇中的一种或两种以上的组合。
根据本发明的相变微胶囊的制备方法,所述水相制备步骤中,所述调节剂包括十二烷基硫醇。
根据本发明的相变微胶囊的制备方法,所述油相制备步骤中,所述芯材包括十八烷。
根据本发明的相变微胶囊的制备方法,所述油相制备步骤中,所述纳米添加剂包括纳米二氧化钛、纳米铜和纳米氧化铝中的一种或两种以上的组合;
优选地,所述纳米添加剂包括所述纳米二氧化钛、纳米铜和纳米氧化铝的组合,更优选地,所述纳米铜的加入量为0.10~0.20重量份;所述纳米二氧化钛的加入量为0.20~0.30重量份;所述纳米氧化铝的加入量为0.10~0.20重量份。
根据本发明的相变微胶囊的制备方法,所述油相制备步骤中,所述引发剂包括偶氮二异丁腈和/或过硫酸钾。
根据本发明所述的相变微胶囊的制备方法,所述破乳步骤中,所述破乳剂包括氯化钙的乙醇溶液,优选地,以所述氯化钙的乙醇溶液的重量百分比计,所述氯化钙的加入量为1~3重量%。
本发明还提供一种根据本发明的制备方法制备得到的相变微胶囊,其特征在于,所述相变微胶囊包括核体和包覆于核体外部的壳体;
所述相变微胶囊的粒径在200~1000nm之间,优选地,60体积%以上的所述相变微胶囊的粒径在400~600nm之间;所述相变微胶囊的相变潜热在80-100j/g之间;所述相变微胶囊的导热系数为0.23~0.28w/m·k,优选0.25~0.28w/m·k。
本发明还提供一种根据本发明的制备方法制备得到的相变微胶囊在皮革制品中的应用。
发明的效果
本发明的相变微胶囊的包封率高和导热性能优异,粒径小,有利于在皮革中进行渗透,较小的粒径也更有利于在皮革加工各相关工序进行应用。进一步地,其能够提高皮革的调温性能,改善皮革的舒适性,提高皮革的使用价值和经济价值。
附图说明
包含在说明书中并且构成说明书的一部分的附图与说明书一起示出了本申请的示例性实施例、特征和方面,并且用于解释本申请的原理。
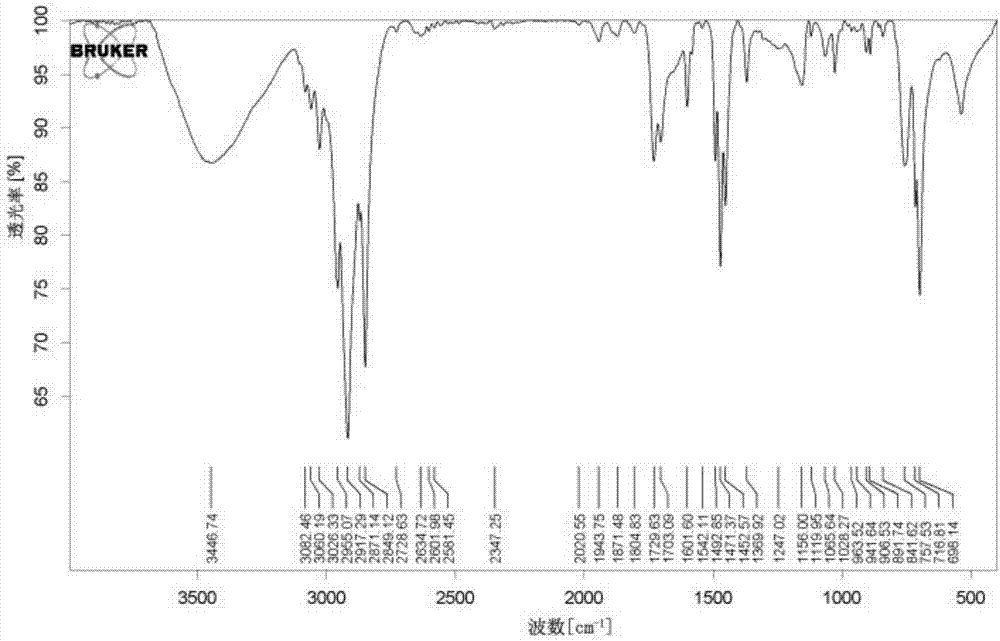
图1示出了本申请实施例1的相变微胶囊的红外光谱图;
图2示出了本申请实施例2的相变微胶囊的红外光谱图;
图3示出了本申请实施例1的相变微胶囊的粒径分布图;
图4示出了本申请实施例2的相变微胶囊的粒径分布图。
具体实施方式
以下将参考附图详细说明本发明的各种示例性实施例、特征和方面。在这里专用的词“示例性”意为“用作例子、实施例或说明性”。这里作为“示例性”所说明的任何实施例不必解释为优于或好于其它实施例。
另外,为了更好地说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在另外一些实例中,对于本领域技术人员熟知的方法、手段、器材和步骤未作详细描述,以便于凸显本发明的主旨。
本发明提供了一种相变微胶囊的制备方法,包括以下步骤:
水相制备步骤:将乳化剂、任选存在的助乳化剂以及任选存在的调节剂溶于水中,分散后得到水相;
油相制备步骤:将单体、芯材、引发剂以及纳米添加剂均匀混合,得到油相;
预乳化步骤:将油相与水相混合,常温下分散并进行预乳化,得到预乳化液;
乳化步骤:将预乳化液进行超声分散,然后在氮气保护下,在75-85℃下加热,反应7-8h后,冷却,得到乳化剂;
破乳步骤:在乳化液中加入破乳剂进行破乳。
在水相制备步骤中,所述乳化剂包括十二烷基硫酸钠和/或烯丙氧基壬基酚聚氧乙烯(10)醚硫酸铵。
所述水相制备步骤中,助乳化剂包括十六烷、十六醇和十八醇中的一种或两种以上的组合。
所述水相制备步骤中,所述调节剂包括十二烷基硫醇。
在本发明的水相制备步骤中,为了有利于各个步骤的有利进行,乳化剂的加入量为1-4重量份,助乳化剂的加入量为0-6重量份,调节剂的加入量为0-3重量份。
一般而言,在水相制备步骤中,在不含有调节剂时,可以将ph调节至中性。例如:可以使用磷酸氢二钠和磷酸二氢铵进行调节,调节ph值在6.5-7.5之间;优选地,可以调节ph在7.0-7.1之间。另外,在本发明中,磷酸氢二钠和磷酸二氢铵的质量比可以为0.5-2:1,优选为1:1。一般而言,在水相制备步骤中,一般采用超声分散的方式,超声分散的时间在10-20分钟之间。
在本发明的油相制备步骤中,所述芯材包括十八烷。
所述油相制备步骤中,所述纳米添加剂包括纳米二氧化钛、纳米铜和纳米氧化铝中的一种或两种以上的组合;通过添加纳米添加剂,可以改善本发明的相变微胶囊的导热性能。
优选地,所述纳米添加剂包括所述纳米二氧化钛、纳米铜和纳米氧化铝的组合,更优选地,所述纳米铜的加入量为0.10~0.20重量份;所述纳米二氧化钛的加入量为0.20~0.30重量份;所述纳米氧化铝的加入量为0.10~0.20重量份。
当使用纳米铜、纳米二氧化钛以及钠米氧化铝的组合时,可以进一步提高相变微胶囊的相变潜热,包封率以及导热系数,进而进一步提高相变微胶囊的导热性能。
所述油相制备步骤中,所述引发剂包括偶氮二异丁腈和/或过硫酸钾。
所述油相制备步骤中,单体可以是苯乙烯、甲基丙烯酸-3-三甲氧基硅丙酯、丙烯酸丁酯和丙烯酸中的一种或两种以上的组合。优选地,苯乙烯与甲基丙烯酸-3-三甲氧基硅丙酯的混合物,或者苯乙烯、丙烯酸丁酯以及丙烯酸的混合物,其中,苯乙烯为主单体。
在本发明的油相制备步骤中,为了有利于各个步骤的有利进行,单体的加入量为20-65重量份;芯材的加入量为15-35重量份;引发剂的加入量为0.5-2重量份。
在本发明的预乳化步骤中,一般可以采用机械分散的方式进行分散,所述机械分散可以采用机械分散器、搅拌器进行分散。一般而言,机械分散的时间为30-40min,此时转速可以为5000-6000r/min。
本发明中,通过预乳化的步骤,可使单体和乳化剂混合十分均匀,使增溶胶束的形成减少,聚合反应变得平稳易于控制。预乳化还可使得乳液的外观、粒径及分布更容易控制,为乳化步骤中的细乳化反应进而制备相变微胶囊提供有利的基础。
在本发明的乳化步骤中,可以将预乳化液继续在5000-6000r/min的转速下进行机械分散10-20min进行乳化,然后进行超声分散20-30min进行细乳化;也可以直接将预乳化液进行超声分散10-20min后,再进行机械分散10-20min,然后再进行超声分散10-30min;还可以直接只进行超声分散。
进一步地,在氮气保护下,在70-85℃下进行加热,反应6-8h后,于室温下自然冷却,得到乳化液。
所述破乳步骤中,所述破乳剂为氯化钙的乙醇溶液,优选地,以所述氯化钙的乙醇溶液的质量百分比计,所述氯化钙的加入量为1-3重量%。
另外,经破乳后,可以再进行清洗,优选地,可以采用乙醇的水溶液清洗1-5次,然后再采用蒸馏水清洗1-5次。更优选地,乙醇的水溶液中,乙醇的质量百分比为40-60%。
本发明还提供一种根据本发明的制备方法制备得到的相变微胶囊,所述相变微胶囊包括核体和包覆于核体外部的壳体;
所述相变微胶囊的粒径在200-1000nm之间,优选地,60体积%以上的所述相变微胶囊的粒径在400-600nm之间;所述相变微胶囊的相变潜热在80-100j/g之间;所述相变微胶囊的导热系数为0.23-0.28w/m·k,优选0.25-0.28w/m·k。
本发明制备得到的相变微胶囊有利于在皮革中进行渗透,较小的粒径也更有利于在皮革加工各相关工序进行应用,提高皮革的调温性能,改善皮革的舒适性,提高皮革的利用价值。
另外,本发明还提供一种根据本发明的制备方法制备得到的相变微胶囊在皮革制品中的应用。
实施例
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的符合一定质量标准的常规产品。
实施例1
称得0.15g十二烷基硫酸钠,0.17g烯丙氧基壬基酚聚氧乙烯(10)醚硫酸铵(dns-86),0.17g十六烷(hd),0.16g十二烷基硫醇(ddm)溶于21.17g水中,超声分散10min得到水相。称得5.00g苯乙烯,0.50g丙烯酸丁酯,0.22g丙烯酸,3.05g十八烷,0.01g纳米铜、0.02g纳米二氧化钛,0.01g纳米氧化铝、0.117g偶氮二异丁腈,混合搅拌均匀,得到油相。将油相加入到水相中,室温搅拌40min进行预乳化。将预乳化液超声分散15min后,在5000r/min的转速下搅拌分散15min,继续超声分散20min。于氮气保护下在85℃下加热7h。结束后室温下自然冷却。加入2重量%的氯化钙乙醇溶液破乳,抽滤,用50%乙醇溶液清洗3次,再用蒸馏水清洗3次,35℃烘干保存。
实施例2
将0.60g十六烷,0.20g十二烷基硫酸钠溶于水中,并以磷酸氢二钠和磷酸二氢铵按1:1调节ph为7.0,超声分散15min得到水相。将2.1g苯乙烯,0.7g甲基丙烯酸-3-三甲氧基硅丙酯,2g十八烷、0.02g纳米铜、0.03g纳米二氧化钛,0.02g纳米氧化铝、0.1g偶氮二异丁腈,混合均匀得到油相;将油相加入到水相中,在6000r/min的转速下室温搅拌分散35min进行预乳化;在6000r/min的转速下将预乳化液搅拌分散15min,然后进行超声分散25min。于氮气保护下在70℃下搅拌加热6h。反应结束后室温下自然冷却。冷却结束后加入2%的氯化钙乙醇溶液破乳,抽滤,用50%乙醇溶液清洗3次,再用蒸馏水清洗3次,40℃烘干保存。
实施例3
称得0.20g十二烷基硫酸钠,0.25g烯丙氧基壬基酚聚氧乙烯(10)醚硫酸铵(dns-86),0.22g十六烷(hd),0.21g十二烷基硫醇(ddm)溶于21.2g水中,超声分散10min得到水相。称得4.50g苯乙烯,0.30g丙烯酸丁酯,0.18g丙烯酸,2.85g十八烷,0.02g纳米二氧化钛,0.02g纳米氧化铝、0.117g偶氮二异丁腈,混合搅拌均匀,得到油相。将油相加入到水相中,室温搅拌40min进行预乳化。将预乳化液超声分散15min后,在5000r/min的转速下搅拌分散15min,继续超声分散20min。于氮气保护下在85℃下加热7h。结束后室温下自然冷却。加入2重量%的氯化钙乙醇溶液破乳,抽滤,用50%乙醇溶液清洗3次,再用蒸馏水清洗3次,35℃烘干保存。
实施例4
将0.70g十六烷,0.30g十二烷基硫酸钠溶于水中,并以磷酸氢二钠和磷酸二氢铵按1:1调节ph为7.0,超声分散15min得到水相。将2.2g苯乙烯,0.8g甲基丙烯酸-3-三甲氧基硅丙酯,2g十八烷、0.02g纳米铜、0.03g纳米二氧化钛、0.1g偶氮二异丁腈,混合均匀得到油相;将油相加入到水相中,在6000r/min的转速下室温搅拌分散35min进行预乳化;在6000r/min的转速下将预乳化液搅拌分散15min,然后进行超声分散25min。于氮气保护下在70℃下搅拌加热6h。反应结束后室温下自然冷却。冷却结束后加入2%的氯化钙乙醇溶液破乳,抽滤,用50%乙醇溶液清洗3次,再用蒸馏水清洗3次,40℃烘干保存。
对比例
称得0.15g十二烷基硫酸钠,0.17g烯丙氧基壬基酚聚氧乙烯(10)醚硫酸铵(dns-86),0.17g十六烷(hd),0.16g十二烷基硫醇(ddm)溶于21.17g水中,超声分散10min得到水相。称得5.00g苯乙烯,0.50g丙烯酸丁酯,0.22g丙烯酸,3.05g十八烷,0.117g偶氮二异丁腈,混合搅拌均匀,得到油相。将油相加入到水相中,室温搅拌40min进行预乳化。将预乳化液超声分散15min后,在5000r/min的转速下搅拌分散15min,继续超声分散20min。于氮气保护下在85℃下加热7h。结束后室温下自然冷却。加入2重量%的氯化钙乙醇溶液破乳,抽滤,用50%乙醇溶液清洗3次,再用蒸馏水清洗3次,35℃烘干保存。
实验效果测试
dsc测试
采用差示扫描量热仪,在n2气氛下,以5℃/min的扫描速率,测量-10℃~80℃范围的dsc曲线,得到反应后相变微胶囊的相变潜热δhw,具体如表1所示。
相变微胶囊的包封率的计算
根据反应后相变微胶囊的相变潜热及反应前芯材(实施例中的十八烷)的质量分数进行计算。具体为:
包封率(%)=反应后相变微胶囊中芯材(实施例中为十八烷)的质量分数(%)/反应前所加入的芯材(实施例中为十八烷)的占总原料的质量分数(%)。
其中,相变微胶囊中芯材(十八烷)的质量分数(%)=δhw/δhp,δhw和δhp分别为dsc测得的反应后相变微胶囊的相变潜热和反应前芯材(实施例的十八烷)的相变潜热(即芯材原料的相变潜热),具体结果如下表1所示。
导热系数测试
采用导热系数测定仪测定导热系数。
表1
由表1可以看出,本申请的实施例1-4的相变微胶囊相比对比例的相变微胶囊,其相变潜热高,包封率高,且导热系数更高。
红外测试
利用bruker红外光谱仪测试实施例1和实施例2的红外光谱,具体如图1和图2所示。图1示出了本申请实施例1的相变微胶囊的红外光谱图;图2示出了本申请实施例2的相变微胶囊的红外光谱图。
由图1可以看出,1050.23cm-1处为si-o-si特征吸收峰,表明合成反应生成了目标产物,生成了si-o-si无机网络结构。2923.17cm-1处和2852.73cm-1处的吸收峰分别是由十八烷中的亚甲基(-ch2)不对称伸缩振动和对称伸缩振动产生的。可见本申请实施例1形成了以十八烷为芯材的相变微胶囊。
由图2可以看出,1703.09cm-1、1729.63cm-1处为羧酸中c=o的特征吸收峰,3446.74cm-1为羧酸中-oh的特征吸收峰,表明合成产物中含有羧基,证实苯乙烯与丙烯酸进行反应生成了产物。716.81cm-1处的吸收峰是由十八烷中4个以上亚甲基(-ch2)相连时水平摇摆振动产生的。1471.37cm-1处的吸收峰是由十八烷中甲基(-ch3)不对称变形振动产生的,2955.07cm-1处的吸收峰是由十八烷中甲基(-ch3)不对称伸缩振动产生的,2917.29cm-1处和2849.12cm-1处的吸收峰分别是由十八烷中亚甲基(-ch2)不对称伸缩振动和对称伸缩振动产生的。由此可见,本申请实施例2形成了以十八烷为芯材的相变微胶囊。
粒径测试
采用激光散射粒度分析仪,测试实施例1和实施例2相变微胶囊的粒径大小及分布。图3示出了本申请实施例1的相变微胶囊的粒径分布图;图4示出了本申请实施例2的相变微胶囊的粒径分布图。
由图3可以看出,实施例1制备得到的相变微胶囊的粒径分布较为集中,大多集中在400-600纳米之间,且500纳米左右的粒径含量较多,集中在400-600纳米之间的粒径在70体积%以上。
由图4可以看出,实施例1制备得到的相变微胶囊的粒径分布较为粒径分布集中且均匀,大多集中在400-600纳米之间,且500纳米左右的粒径含量较多,集中在400-600纳米之间的粒径在80体积%以上。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!