一种亲水性蒽类荧光染料及其合成方法与流程
[0001]
本发明涉及一种亲水性蒽类荧光染料及其合成方法,属于生物化学类荧光染料合成领域。
背景技术:
[0002]
蒽类荧光染料属于一类含有类蒽环的荧光素。该类化合物部分具有长波长、荧光切换、摩尔吸收系数高等特点。这类化合物尤其可以使用在基因测序方面。申请人之前的专利cn201510155218.9中也公开了蒽类化合物有关合成方法。在染料测序实验中发现,原有的荧光染料水溶性比较差,量子效率相较于氧杂蒽类化合物有所降低;为了改善染料的水溶性和提高染料的光学稳定性;特别开发一种含有双氟和磺酸根的荧光染料。进一步发现此染料引入氟原子后,荧光量子产率有所增强;在溶液中较远原来的染料更加不易被光淬灭,稳定性进一步增加。在引入磺酸基团后,染料的水溶性明显改善。
技术实现要素:
[0003]
本发明的目的是通过以下技术方案实现的。本发明提供一种亲水性蒽类荧光染料,其特征在于具有如下所述结构
[0004][0005]
根据优选的实施方式,所述的荧光染料用于基因测序中的碱基标记或者细胞荧光染色。
[0006]
本发明公开一种亲水性蒽类荧光染料的合成方法,其特征在于包括,
[0007]
1)卤代苯甲醛与苯基格氏试剂反应,生成二苯甲醇衍生物,即第一中间产物;
[0008]
2)将第一中间产物的醇羟基氧化为酮羰基,得到带有酚羟基保护基的二苯甲酮衍生物,即第二中间产物;
[0009]
3)将第二中间产物酮羰基用乙二硫醇转化为二硫缩酮进行保护,即第三中间产物;
[0010]
4)将第三中间产物用氟试剂进行氟化获得二氟化蒽酮类化合物,即第四中间产物;
[0011]
5)将第四中间产物与锂试剂反应转化为锂格氏试剂,锂试剂与邻甲基苯甲醛反应生成苯甲醇类衍生物,即第五中间产物;
[0012]
6)将第五中间产物苯甲醇氧化为苯甲酮,即第六中间产物;
[0013]
7)将第六中间产物苯甲酮脱掉保护基,酸性条件下环化反应,即第七中间产物;
[0014]
8)将第七中间产物氟化荧光染料进行磺酸化反应,获得氟代磺酸化荧光染料;
[0015]
根据优选的实施方式,所述步骤1)中卤代苯甲醛与苯基格氏试剂反应指的是,2-溴-5-甲氧基苯甲醛与3-甲氧基苯基格氏试剂反应。
[0016]
根据优选的实施方式,所述步骤4)中的氟试剂,指的是氟化氢吡啶与选择性氟试剂。
[0017]
根据优选的实施方式,所述步骤7)中将第六中间产物苯甲酮脱掉保护基,酸性条件下环化反应,指的是将第六中间产物二苯基甲酮衍生物用三溴化硼脱掉酚羟基保护基,甲基磺酸为溶剂加热条件下进行分子内环化反应,生成氟代蒽类荧光染料。
[0018]
本发明还公开一种亲水性蒽类荧光染料的合成方法,其特征在于包括,
[0019]
1)2-溴-5-甲氧基苯甲醛与3-甲氧基苯基格氏试剂反应,生成二间甲氧基二苯基甲醇衍生物,即第一中间产物;
[0020]
2)将第一中间产物的二间甲氧基二苯基甲醇的醇羟基氧化为酮羰基,得到二间甲氧基二苯基甲酮衍生物,即第二中间产物;
[0021]
3)将第二中间产物二间甲氧基二苯基甲酮的酮羰基用乙二硫醇转化为二间甲氧基二苯基二硫缩酮进行保护,即第三中间产物;
[0022]
4)将第三中间产物二间甲氧基二苯基二硫缩酮用氟试剂(氟化氢吡啶与选择性氟试剂selecetfluor)进行氟化反应获得二间甲氧基二苯基二氟化甲烷衍生物,即第四中间产物;
[0023]
5)将第四中间产物二间甲氧基二苯基二氟化甲烷衍生物与正丁基锂试剂反应转化为锂格氏试剂,锂格式试剂与邻甲基苯甲醛反应生成二苯基甲醇类衍生物,即第五中间产物;
[0024]
6)将第五中间产物二苯基甲醇类衍生物用氧化剂重铬酸吡啶盐把醇羟基氧化为二苯基甲酮衍生物,即第六中间产物;
[0025]
7)将第六中间产物二苯基甲酮衍生物用三溴化硼脱掉酚羟基保护基,甲基磺酸为溶剂加热条件下进行分子内环化反应,生成氟代蒽类荧光染料;即第七中间产物;
[0026]
8)将第七中间产物氟化荧光染料在浓硫酸中进行磺酸化反应,获得氟代磺酸基蒽类荧光染料。
[0027]
本发明公开一种新型含氟磺化蒽类荧光染料,其具备以下特点:激发波长为近红外,激发波长范围在460-540nm,优选506-521nm;发射波长范围在510-610,优选530-550nm,量子产率为60%左右。同之前公开的专利中的染料相比,引入双氟原子和磺酸根,即改善了其水和脂溶性,便于通过细胞壁,有改善了水溶性便于细胞内蛋白染色标记。
[0028]
本发明公开了一种含氟磺酸类蒽类荧光染料的合成方法,具备以下的优点:
[0029]
(1)提高了染料的水溶性:相比于专利cn111205669a说明书第0085段所述的染料,其特点为:该荧光染料在中性缓冲溶液中溶解度显著增加,较以前荧光染料在中性缓冲溶液中溶解度增加40%,对于测定荧光染料的各种光学性质更加方便。
[0030]
(2)提高了稳定性:引入氟原子后,含氟荧光染料光学稳定性增加,不容易被光淬灭;自身稳定性增加,不会因光照产生自由基从而破坏染料的自身结构和性质,储存时间长,保管容易。
[0031]
(3)由于染料分子含有氟原子,而氟原子具有高电负性和较小的原子半径并且具
有特别低的可极化性;从而使含氟染料具有一些特殊的性质,如含氟化合物更容易通过脂溶性细胞膜,可以用于细胞内dna的染色剂。
[0032]
(4)该染料的波长较氧杂蒽类染料激发和发射发生了红移,但是比其它碳杂蒽类和硅杂蒽类化合物发生蓝移,从而拓宽染料使用的可选择范围。
[0033]
说明书附图
[0034]
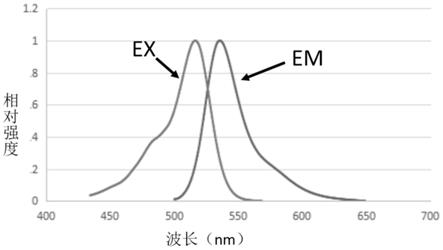
图1.染料的吸收发射光谱。
具体实施方式
[0035]
下面更详细地描述本公开的示例性实施方式。应当理解的,可以以各种形式实现本公开而不应被这里阐述的实施方式所限制。相反,提供这些实施方式是为了能够更透彻地理解本公开,并且能够将本公开的范围完整的传达给本领域的技术人员。
[0036]
除特殊解释外,本发明所涉及的词语均为本领域的常见词语。
[0037]
本发明中所涉及的蒽类化合物,指的是具有类似三苯环蒽结构的化合物。该词语并非特殊用语。
[0038]
本发明所涉及的反应中,某些步骤存在其他产物,但是在本发明所做说明的基础上,本领域技术人员很容易获得目标产物。副产物的具体结构不属于本发明的核心内容,不进行过多描述。
[0039]
下面以具体的路线例子来更进一步的说明本发明。参见下面合成路线1:
[0040][0041]
荧光染料的合成见合成路线:
[0042][0043]
1化合物(2-bromo-5-methoxyphenyl)(3-methoxyphenyl)methanol 3的合成:
[0044]
将化合物1(10g,46.7mmol),溶于无水的四氢呋喃溶液中,冰浴降温至零度,慢慢滴加格氏试剂2(9.8g,46.7mmol),反应2小时。待反应完全后向反应体系中加入少量水淬灭反应,加入稀盐酸水溶液,二氯甲烷萃取,旋除溶剂柱层析纯化可得油状化合物3(39.6g,yield 85%)。
[0045]
1h nmr(300mhz,cdcl3)δ7.45(d,j=7.5hz,1h),7.27(t,j=7.5hz,1h),7.19(dq,j=7.5,1.4hz,1h),7.00
–
6.92(m,2h),6.90
–
6.83(m,2h),6.09(dq,j=6.8,1.0hz,1h),4.69(d,j=6.8hz,1h),3.83(d,j=5.7hz,6h).13c nmr(75mhz,cdcl3)δ159.46,158.43,140.13,138.36,133.25,129.14,122.86,117.09,115.08,113.52,113.47,112.79,74.00,55.62,55.28.lcms:c15h15bro3(m+h),323.1860。
[0046][0047]
2化合物(2-bromo-5-methoxyphenyl)(3-methoxyphenyl)methanone 4合成:
[0048]
将化合物3(12g,37.2mmol)溶于二氯甲烷溶液200ml中,加入重铬酸吡啶盐(16g,74.4mmol),加入硅藻土或硅胶粉(16g),室温反应3小时,tcl监测反应进行至完全,硅藻土抽滤除去不溶物,二氯甲烷洗涤两遍,,旋除溶剂,柱层析分离可得油状化合物4(11g,yield 91%)。
[0049]
1h nmr(300mhz,cdcl3)δ7.66(d,j=7.5hz,1h),7.50(dt,j=7.5,1.6hz,1h),7.41(t,j=7.5hz,1h),7.33(t,j=1.5hz,1h),7.18(dt,j=7.5,1.5hz,1h),7.15(d,j=1.5hz,1h),6.95(dd,j=7.5,1.5hz,1h),3.83(d,j=5.9hz,6h).13c nmr(75mhz,cdcl3)δ194.25,159.52,158.85,137.02,136.84,133.35,128.61,123.38,117.70,116.89,115.59,114.06,114.03,55.77,55.45.lcms:c15h14bro3(m+h),321.0048。
[0050][0051]
3化合物2-(2-bromo-5-methoxyphenyl)-2-(3-methoxyphenyl)-1,3-dithiolane 5合成:
[0052]
将化合物4(9g,28.1mmol)溶于二氯甲烷溶液56mml中,氩气保护下降温至0℃,加入乙二硫醇(4.5ml,28.1mmol)与上述混合溶液中,慢慢滴加三氟化硼乙醚溶液(9.8ml,56.2mmol)反应8小时,将反应体系倒入水溶液中,碳酸氢钠调节ph值至中性后,乙酸乙酯萃取,旋除溶剂纯化可得化合物5(9.5g yield 83%)。
[0053]
1h nmr(300mhz,cdcl3)δ7.42(d,j=7.5hz,1h),7.29(t,j=7.4hz,1h),7.17(dt,j=7.5,1.5hz,1h),7.03(t,j=1.5hz,1h),6.88(d,j=1.5hz,1h),6.84(dt,j=7.5,
1.6hz,1h),6.79(dd,j=7.5,1.5hz,1h),3.81(d,j=4.9hz,6h),3.53
–
3.41(m,4h).13c nmr(75mhz,cdcl3)δ160.72,160.20,141.39,140.22,133.46,128.13,122.48,118.71,115.44,114.85,112.39,111.91,75.70,55.61,55.19,39.31.
[0054]
lcms:c17h18bro2s2(m+h),398.3450。
[0055][0056]
4化合物1-bromo-2-(difluoro(3-methoxyphenyl)methyl)-4-methoxybenzene 6合成:
[0057]
在塑料反应瓶中加入氟试剂(4.3g,12.2mmol)和氟化氢吡啶溶液3ml,氩气保护下降温至0℃搅拌反应15分钟,将化合物5(2.5g,6.2mmol)溶于无水二氯甲烷20ml中,慢慢滴加到氟化氢吡啶溶解的氟试剂溶液中反应1小时,将反应体系倒入水溶液中,碳酸氢钠调节ph值至中性后,乙酸乙酯萃取,旋除溶剂纯化可得化合物6(1.3g,yield 62%)。
[0058]
1h nmr(300mhz,cdcl3)δ7.54(d,j=7.5hz,1h),7.41
–
7.34(m,1h),7.34(dt,j=7.5,1.6hz,1h),7.10(d,j=1.5hz,1h),7.01(t,j=1.5hz,1h),6.95(dt,j=7.1,1.6hz,1h),6.86(dd,j=7.5,1.7hz,1h),3.83(d,j=1.5hz,6h).13c nmr(75mhz,cdcl3)δ159.23,156.15,137.86,137.77,137.60,137.51,137.35,137.26,134.96,129.88,124.61,124.58,124.55,119.74,117.60,115.45,114.94,114.67,114.64,114.61,113.92,113.88,113.85,113.60,113.22,113.19,113.16,55.63,55.22.lcms:c15h14bro2f2(m+h),343.1678。
[0059][0060]
5化合物(2-(difluoro(3-methoxyphenyl)methyl)-4-methoxyphenyl)(o-tolyl)methanol 8合成:
[0061]
将化合物6(4g,11.7mmol)溶于60ml无水的四氢呋喃溶液中,降温至-78℃后加入正丁基锂4.8ml,在此温度下继续反应30分钟;然后将溶有化合物7的四氢呋喃溶液慢慢加入到上述反应体系中,慢慢升至室温继续反应2小时后;tcl检测反应情况,待反应完全后加入稀盐酸5ml,水20ml继续搅拌反应10分钟;旋除溶剂,乙酸乙酯萃取柱层析纯化可得油状化合物8(4.3g yield 73%)。
[0062]
1h nmr(300mhz,cdcl3)δ7.41
–
7.33(m,3h),7.30(dt,j=7.5,1.5hz,1h),7.20(dtd,j=18.3,7.3,1.7hz,2h),7.13(ddt,j=7.2,1.9,0.8hz,1h),7.09(d,j=1.5hz,1h),7.03(t,j=1.5hz,1h),6.93(dt,j=7.3,1.5hz,1h),6.79(dd,j=7.4,1.5hz,1h),6.17(dt,j=6.1,0.9hz,1h),3.94(d,j=5.9hz,1h),3.83(d,j=5.7hz,6h),2.29(d,j=0.7hz,3h).13c nmr(75mhz,cdcl3)δ158.69,158.29,141.22,138.62,138.36,138.11,137.03,136.78,136.52,136.17,133.85,133.82,133.78,129.91,129.55,129.51,127.74,127.28,127.18,121.81,121.78,121.74,120.12,117.97,115.83,113.72,113.17,112.97,112.93,
112.90,112.45,112.42,112.39,69.06,55.60,55.19,19.73.lcms:c23h23f2o3(m+h),385.1571.
[0063][0064]
6化合物(2-(difluoro(3-methoxyphenyl)methyl)-4-methoxyphenyl)(o-tolyl)methanone 9合成:
[0065]
将化合物8(4.4g,11.4mmol)溶于二氯甲烷溶液20ml中,加入重铬酸吡啶盐(6.2g,28.5mmol),加入硅藻土或硅胶粉6g,室温反应3小时,tcl监测反应进行至完全,硅藻土抽滤除去不溶物,二氯甲烷洗涤两遍,旋除溶剂,柱层析分离可得油状化合物9(3.4g,yield 89%)。
[0066]
1h nmr(300mhz,cdcl3)δ7.90(dd,j=7.5,1.6hz,1h),7.59(d,j=7.4hz,1h),7.42(td,j=7.4,1.6hz,1h),7.36(t,j=7.4hz,1h),7.36
–
7.29(m,2h),7.28(ddd,j=7.4,1.6,0.8hz,1h),7.08
–
7.02(m,2h),6.98
–
6.90(m,2h),3.82(s,6h),2.39(s,2h).13c nmr(75mhz,cdcl3)δ196.29,161.35,158.69,139.37,139.11,138.85,137.21,136.97,136.95,136.70,135.98,130.42,130.01,129.98,129.95,129.92,129.91,129.77,128.64,126.88,121.83,121.80,121.77,119.32,117.18,115.03,113.24,113.17,112.97,112.93,112.90,112.69,112.66,112.63,55.61,55.19,19.74.lcms:c23h21f2o3(m+h),383.1381.
[0067][0068]
7化合物9,9-difluoro-7-hydroxy-10-(o-tolyl)anthracen-2(9h)-one 10合成:
[0069]
将化合物9(3.3g,8.8mmol)溶于50ml无水的二氯甲烷溶液中,降温至0℃后加入三溴化硼(3ml,31.7mmol),升至室温下继续反应3小时;然后加入水淬灭,二氯甲烷萃取,水洗后有几层溶剂干燥,除去溶剂后,加入甲基磺酸6ml,加热至90℃反应8小时,将反应物倒入冰水中,乙酸乙酯萃取柱层析纯化可得黑色固体化合物10(1.8g yield 60%)。
[0070]
1h nmr(300mhz,cdcl3)δ8.99(s,1h),7.53(d,j=7.5hz,1h),7.44(d,j=10.8hz,1h),7.40
–
7.32(m,2h),7.26
–
7.17(m,2h),7.01(d,j=1.5hz,1h),6.88(d,j=2.0hz,1h),6.80(dd,j=7.5,1.5hz,1h),6.60(dd,j=10.9,1.9hz,1h),2.40(d,j=0.7hz,3h).13c nmr(75mhz,cdcl3)δ186.36,158.17,139.94,139.68,139.43,139.13,138.35,136.76,134.16,133.65,133.56,133.53,133.50,133.40,133.14,132.13,129.05,128.63,128.19,127.25,127.22,127.19,126.97,126.39,121.44,121.40,121.37,115.66,113.63,113.11,113.08,113.05,111.49,109.34,20.43.lcms:c21h15f2o2(m+h),337.0962。
[0071][0072]
8化合物3-(10,10-difluoro-6-hydroxy-3-oxo-3,10-dihydroanthracen-9-yl)-4-methylbenzenesulfonic acid 11合成:
[0073]
化合物10(1.0g,2.9mmol)溶于5ml浓硫酸中,室温下反应2小时,将反应溶液倒入冰水混合物中静止2小时;抽滤溶液中红色沉淀,乙酸乙酯冲洗两遍,可得纯的红色固体化合物11(1.1g yield 92%)。
[0074]
1h nmr(300mhz,meod)δ9.58(s,1h),8.97(s,1h),8.04(d,j=1.5hz,1h),7.65(dd,j=7.5,1.5hz,1h),7.61
–
7.53(m,2h),7.51(dq,j=7.5,1.0hz,1h),7.04(d,j=1.5hz,1h),6.92(d,j=1.8hz,1h),6.81(dd,j=7.4,1.5hz,1h),6.62(dd,j=10.9,1.9hz,1h),2.33(d,j=1.0hz,3h).13c nmr(75mhz,meod)δ185.95,156.55,139.56,139.40,139.15,138.89,138.68,137.90,137.74,134.14,133.95,133.69,133.44,133.15,133.11,133.08,132.02,130.96,129.14,126.97,126.58,126.17,126.14,126.10,121.54,121.51,121.48,115.80,113.63,112.90,112.87,112.83,111.49,109.34,20.64.lcms:c21h15f2o5s(m+h),417.0530。
[0075]
化合物11的荧光吸收发射光谱数据参见图1。相比于专利,染料吸收发射光谱都发生了变化,具体的之前专利cn111205669a中化合物0062,发射光谱范围约为:510-590nm;吸收光谱范围约为:550-650nm。目前染料的发射光谱范围约为:460-540nm吸收光谱范围约为:510-610nm。
[0076]
化合物11的亲水性得到了明显的改善。专利cn111205669a的化合物0062,水溶性为:2mg/10ml。化合物11的水溶性为:7mg/10ml。该染料主要用于基因测序或者生物标记,水溶性的提高可以明显的拓宽使用的范围,降低使用条件需求。
[0077]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1