一种具有高韧性高致密性网状交联结构壳材的相变微胶囊及其制备方法与流程
1.本发明属于精细化工和轻化工技术领域,具体涉及一种具有高韧性高致密性网状交联结构的微胶囊型相变材料及其制备方法。
背景技术:
2.相变微胶囊材料(pcm-phase change microcapsule material,pcmm),是一种具有特定功能的物质,它能在特定温度(相变温度)下发生物相变化,并伴随吸收或释放热能的现象来贮存或放出热能,进而调整、控制工作源或材料周围环境温度,以实现其特定的应用功能。近年来,相变微胶囊材料发展迅速,可广泛应用于建筑、服装和医护等领域。包覆相变材料的微胶囊主要采用三聚氰胺甲醛树脂和聚氨酯脲树脂为壳材,由于前者不可避免的存在游离甲醛问题,不利于环境保护。
3.中国发明专利cn03116242以一种利用双或多异氰酸酯化合物与小分子胺类交联反应作为形成聚氨酯脲微胶囊壳材。但双异氰酸酯化合物与小分子胺类反应形成的壳材多为线性结构,多异氰酸酯与小分子胺类交联没有引入柔性链段,致使所制备的微胶囊的致密性和韧性差。另外,由于微胶囊型相变材料经过长期吸热和放热循环后芯材易泄露,聚氨酯脲微胶囊型相变材料的适用性也受到了一定限制。
4.中国专利cn110627996a公开了一种聚氨酯脲、其制备方法及基于其的超强韧聚氨酯脲,具有较高的机械强度,用作医学材料。中国专利cn110527060a公开了一种聚氨酯脲组合物及其聚氨酯脲涂层制备方法,克服现有热熔胶型轮胎内层低温硬化、且污染环境的问题。虽然提高了聚氨酯脲的致密性,但体系中仍然具有较多的游离异氰酸根,聚氨酯脲的致密性仍有上升的空间。
技术实现要素:
5.本发明的目的是针对现有技术的不足,本发明以聚氨酯脲为微胶囊的壳材,以正烷烃为微胶囊型的芯材,采用界面聚合法得到具有高韧性和致密性网状交联结构壳材的微胶囊,壳材致密性、韧性和热稳定性好,粒径可控,相变潜热值高,在纺织品,冷链和航空航天等领域具有广阔的发展前景。
6.为实现上述目的,本发明采用的技术方案如下:
7.本发明首先提供一种具有高韧性高致密性网状交联结构壳材的相变微胶囊的制备方法,包括以下步骤:
8.1)配制水相:配制乳化剂水溶液,得到水相;
9.2)配制油相:二异氰酸酯与官能团数为3~4的聚醚多元醇反应3~5小时,得到多个nco端基的聚氨酯半预聚体混合物,其中二异氰酸酯的-nco与聚醚多元醇的-oh的摩尔比为5:1~10:1;芯材正烷烃和多个nco端基的聚氨酯半预聚体混合物混合均匀,得到油相;
10.3)乳化:在高速搅拌下将油相分散于水相,加入乳化剂,乳化时间1~20min,温度
40~90℃,形成稳定的水包油型乳液;
11.4)胶囊化:在40~90℃搅拌下,加入多官能团胺类交联剂,反应0.5~1小时后,加入小分子胺类交联剂,保温0.5~2小时。升温至90~150℃后保温2~7小时;
12.5)出料,洗涤,制得聚氨酯脲壳材的相变微胶囊粉。
13.具体的,所述二异氰酸酯为甲苯二异氰酸酯(tdi)、异佛尔酮二异氰酸酯(ipdi)、二苯基甲烷二异氰酸酯(mdi)、二环己基甲烷二异氰酸酯(hmdi)、六亚甲基二异氰酸酯(hdi)、赖氨酸二异氰酸酯(ldi)中的一种。
14.具体的,所述官能团数为3~4的聚醚多元醇为三羟基丙烷聚醚多元醇或季戊四醇聚醚多元醇或两者的混合物;
15.进一步的,所述官能团数为3~4的聚醚多元醇为聚醚多元醇204,聚醚多元醇220和聚醚多元醇306中的一种或几种。
16.所述多个nco端基的聚氨酯半预聚体混合物为多个nco端基的聚氨酯半预聚体和未反应完的二异氰酸酯的混合物。
17.油相中的芯材与聚氨酯半预聚体混合物的比例影响微胶囊壳材的厚度。具体的,所述芯材正烷烃与多个nco端基的聚氨酯半预聚体混合物的质量比为1~7:1。
18.进一步的,所述乳化剂可以是非离子型的,也可以是阴离子的,其亲水亲油平衡值(hlb)最好在10~16之间。如聚乙二醇醚600、聚醚2040、聚醚f-68等非离子表面活性剂,苯乙烯-马来酸酐共聚物、十二烷基苯磺酸钠和十二烷基硫酸钠等阴离子表面活性剂或它们适当的混合物;乳化剂用量为总体系质量的0.1%至5.0%。
19.进一步的,在步骤4)将催化剂加入体系,目的在于去除游离的异氰酸根基团,所述的催化剂是二月桂酸二丁基锡,用量为总体系重量的0%至0.01%。
20.在步骤4)中加入催化剂二月桂酸二丁基锡,用量为总体系质量的0%至0.01%。
21.具体的,所述多官能团胺类交联剂为聚乙烯亚胺或壳聚糖;小分子胺类交联剂为具有rnnhm的分子通式,其中r=c1~c4的烷基,n=0~2,m=3~1。
22.优选的,小分子胺类交联剂为尿素、丙二胺、二乙烯三胺和丁二胺。
23.先加入多官能团胺类交联剂,形成交联网络结构。反应0.5-1小时后,再加入更容易扩散到界面中的小分子胺类,与剩余的异氰酸酯基反应,以进一步提高壳材致密性。
24.具体的,多官能团胺类交联剂和小分子胺类交联剂用量均为总体系质量的0.005%~5%。
25.本发明的设计思路包括三步:(1)形成多个端基nco的聚氨酯半预聚体;
26.以3官能团的聚醚多元醇为例,与二异氰酸酯反应,形成3个端基nco的聚氨酯半预聚体,方程式如下:
[0027][0028]
(2)多个nco端基的聚氨酯半预聚体与多官能团的胺类反应形成交联网络结构;以
3个nco端基的聚氨酯半预聚体与聚乙烯亚胺反应为例,其方程式如下:
[0029][0030]
(3)在体系中加入小分子交联剂后,以丙二胺或尿素,或者丙二胺与尿素复配为例,其反应方程式如下:
[0031][0032]
本发明还提供一种具有高韧性高致密性网状交联结构壳材的相变微胶囊,由上述任一项制备方法制得。
[0033]
与现有技术相比,本发明具有如下突出效果:
[0034]
1)通过二异氰酸酯与官能团数为3~4的聚醚多元醇反应,多官能团提高交联度,增加致密性,聚醚多元醇的长链段提供韧性;另外考虑官能度和黏度的平衡,聚醚多元醇和二异氰酸酯的比例也需控制在一定范围内,防止入过多的聚醚多元醇,导致预聚物黏度太高,难以与烷烃混合乳化;
[0035]
2)加入小分子胺是因为在水相中聚合物胺的扩散会在一定交联度后会受到抑制,需要更容易扩散的小分子胺进去反应来提高致密性。而如果将聚合物胺即多官能团的胺类交联剂和小分子胺类交联剂复配一起加入,由于小分子胺反应更快,聚合物胺难以扩散,难以形成更高交联度的聚脲产物;
[0036]
3)通过上述方法制备得到的微胶囊具有高韧性和致密性网状交联结构的聚氨酯脲壳材,使用的助剂少,制备简单,成本低;该微胶囊无甲醛,无污染,抗渗透,机械强度好,过长期吸热和放热循环后,还能保持芯材不泄露。
附图说明
[0037]
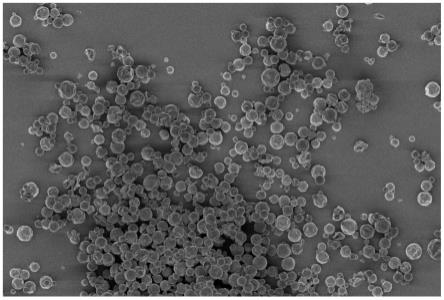
图1为实施例1制备得到的聚氨酯脲相变微胶囊的sem图;
[0038]
图2为实施例1制备得到的聚氨酯脲相变微胶囊聚氨酯脲相变微胶囊经50次冷热循环后的dsc曲线。
具体实施方式
[0039]
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进。这些都属于本发明的保护范围。
[0040]
实施例1
[0041]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0042]
2)油相配制:称取2g ipdi,0.3g聚醚多元醇204和0.3g聚醚多元醇306,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0043]
称取正十八烷12g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0044]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0045]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时后,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为10.36μm的聚氨酯脲微胶囊粉末,观察的sem图如图1所示。制备得到的聚氨酯脲相变微胶囊的50次冷热循环测试前后的dsc结果如图2所示。
[0046]
实施例2
[0047]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0048]
2)油相配制:称取2g ipdi,0.5g季戊四醇,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0049]
称取正十八烷12g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0050]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0051]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时后,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为9.81μm的聚氨酯脲微胶囊粉末。
[0052]
实施例3
[0053]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0054]
2)油相配制:称取2g ipdi,0.5g聚醚多元醇204和0.4g聚醚多元醇306,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0055]
称取正十八烷12g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0056]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0057]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为11.02μm的聚氨酯脲微胶囊粉末。
[0058]
实施例4
[0059]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0060]
2)油相配制:称取2g ipdi,0.25g聚醚多元醇204和0.2g聚醚多元醇306,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0061]
称取正十八烷12g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0062]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0063]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为12.35μm的聚氨酯脲微胶囊粉末。
[0064]
实施例5
[0065]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0066]
2)油相配制:称取2g ipdi,0.3g聚醚多元醇204和0.3g聚醚多元醇306,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0067]
称取正十八烷15g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0068]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0069]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为10.57μm的聚氨酯脲微胶囊粉末。
[0070]
实施例6
[0071]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0072]
2)油相配制:称取2g ipdi,0.3g聚醚多元醇204和0.3g聚醚多元醇306,70℃反应1小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0073]
称取正十八烷5g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0074]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0075]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙烯三胺,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为9.87μm的聚氨酯脲微胶囊粉末。
[0076]
对比例1
[0077]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0078]
2)油相配制:称取正十八烷12g和2g ipdi在100℃混合均匀,得到油相。
[0079]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0080]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g聚乙烯亚胺。保温1小时后升温至80℃,加入0.5g二乙稀三胺,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为10.46μm的聚氨酯脲微胶囊粉末。
[0081]
对比例2
[0082]
1)水相的配制:称取3%苯乙烯-马来酸酐共聚物1.5g溶于烧杯中,加入180g去离子水,搅拌,使乳化剂完全溶解,得到水相。
[0083]
2)油相配制:称取2g ipdi,0.3g聚醚多元醇204和0.3g聚醚多元醇306,70℃反应3小时,得到多个nco端基的聚氨酯半预聚体混合物;
[0084]
称取正十八烷12g和多个nco端基的聚氨酯半预聚体混合物在100℃混合均匀,得到油相。
[0085]
3)乳液的形成:将水相在50℃下保温5分钟后,将油相加入到水相中,4000rpm高速分散20分钟,形成稳定的水包油型乳液;
[0086]
4)微胶囊的形成:乳化结束后,降至2000rpm转速,向上述乳液加入1.5g二乙烯三胺。保温1小时后升温至80℃,保温1小时候,升温至90℃继续保温5小时,结束反应,得到高韧性和致密性网状交联结构聚氨酯脲微胶囊乳液。将乳液用适当去离子水稀释,静置陈化12小时后,取上层微胶囊饼,用去离子水分散,减压抽滤,得到滤饼。滤饼分散于去离子水中后,减压抽滤,洗涤3次。置于70℃烘箱中干燥5小时,得到粒径为11.84μm的聚氨酯脲微胶囊粉末。
[0087]
微胶囊耐压强度测试
[0088]
测试方法:在装有套筒的标准压片模具底座平台上铺入合适的称量纸圆片,平台面积为1cm2。称取100mg样品,精确至1mg。轻敲底座基座及套筒侧面,使待测样品均匀铺展在平台上,将另一片称量纸圆片轻轻覆盖在待测样品上,装上模具压顶,轻敲套筒,旋转压顶排出待测样品空气。将整套模具置于推拉力测试台底座中部,轻压下压力测试仪,再次调整模具位置在正中线上压下测试仪并维持压力读数在0.1
±
0.005kn范围内,持续20s后上抬测试仪,取下标准压片模具,将模具倒立,轻轻取下模具底座,观察粉体压片中是否有油滴痕迹。若粉体压片中无油滴痕迹,揭开上部称量纸圆片,观察称量纸及压片上是否存在油渍。
[0089]
若未观察到明显油渍,可以清洁测试台及标准压力模具后重新按上述步骤提高0.05kn压力重复测试,直至出现油渍,并记录未出现油渍时最高压力大小;若观察到明显油渍则清洁测试台及标准压力模具后重新按上述步骤降低0.05kn压力重复测试,直至油渍消失,并记录压力大小。
[0090]
微胶囊耐温测试
[0091]
测试方法:取微胶囊粉末2g至锡箔纸上,置入150℃烘箱1小时后取出。取微胶囊粉
末少量于玻片上展开,在显微镜下观察微胶囊是否粘连。若微胶囊未出现粘连现象,则该工艺制备的微胶囊耐压合格,否则不合格,结果如表1所示。
[0092]
表1 实施例1~6和对比实施例的耐温耐压的测试结果
[0093][0094][0095]
结果显示,实施例1~6中由于多官能团胺类交联剂和小分子胺类交联剂的加入,以及引入聚醚多元醇,制备得到的聚氨酯脲微胶囊壳材的韧性和致密性更好,耐压性能优异。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1