一种利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法与流程
[0001]
本发明属于水污染控制技术领域,涉及一种利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法。
背景技术:
[0002]
随着工业化的不断发展,产生了越来越多的有机污染物,这些有机污染物存在于环境中,对于整个生态环境造成了极大的危害,因而有机污染水体受到了越来越多的关注。以四环素(tc)为例,它是最广泛用于生长生产和疾病控制的抗生素。由于tc在人和牲畜中的吸附和代谢较差,因此30-90%的tc及其代谢产物通过粪便和尿液排出。地表水和地下水中的tc浓度通常在μg/l到mg/l之间,这对人类健康和生态系统构成威胁。尤其是环境中的tc残留物会改变微生物群落的多样性和组成,从而导致tc耐药细菌和基因的出现。迄今为止,已经探索了包括吸附、生物降解、电解和光催化在内的各种技术,但是效率不佳或高能量输入限制了它们的大规模应用。因此,迫切需要开发一种高效、低成本的方法来消除水中的tc。
[0003]
高级氧化技术(aops)已被公认为是降解难降解有机污染物的一种有前途的方法。其中,过氧化氢、过二硫酸盐和过一硫酸盐(pms)是用于aops的常见氧化剂。与其他两种氧化剂相比,pms具有更长的过氧键和不对称结构,使其更易于离解。然而,pms本身氧化有机污染物的能力有限,因此需要通过其他方法(例如,紫外线照射、加热、超声和添加催化剂)进行刺激以生成高反应性物质,例如硫酸根自由基(so4·-)、羟基自由基(
·
oh)和单线态氧(1o2),即“pms活化”。例如,包括浸渍钴的生物炭、零价铁和锰铁双金属氧化物等在内的催化剂均有效活化pms以通过产生so4·-来氧化降解tc。尽管在这些研究中已经达到令人满意的降解效率,但是反应介质是在超纯水中进行的,并且由于自由基清除作用,tc降解被水组分(例如天然有机物,卤素阴离子和碳酸氢根离子)显著抑制。而且,在实际应用之前,仍需存在有毒金属离子浸出的风险。
技术实现要素:
[0004]
本发明需要解决的技术问题是克服现有技术的不足,提供一种处理成本低、抗水基质干扰能力强、降解效率高、绿色环保的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法。
[0005]
为解决上述技术问题,本发明采用的技术方案为:
[0006]
一种利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,包括以下步骤:将磁性氮掺杂碳、过硫酸盐与含有机污染物水体混合进行降解反应,完成对水体中有机污染物的降解;所述磁性氮掺杂碳包括氮掺杂碳,所述氮掺杂碳中包裹有co纳米颗粒。
[0007]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述磁性氮掺杂碳是以zif-67材料为原料经过煅烧和酸刻蚀后制备得到,包括以下
步骤:
[0008]
s1、将zif-67材料进行煅烧,得到黑色产物;
[0009]
s2、将步骤s1中得到的黑色产物浸泡到酸溶液中,清洗,干燥,得到磁性氮掺杂碳。
[0010]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,步骤s1中,所述煅烧在氩气气氛或氮气气氛下进行;所述煅烧过程中的升温速率为2℃/min~5℃/min;所述煅烧的温度为600℃~1100℃;所述煅烧的时间为在2h~4h。
[0011]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,步骤s1中,所述zif-67材料由以下方法制备得到:将含钴溶液与2-甲基咪唑溶液混合,搅拌8h~24h,离心,采用甲醇洗涤离心后的产物3次~5次,在40℃~70℃的真空条件下干燥,得到zif-67材料;所述含钴溶液中的钴离子与2-甲基咪唑溶液中的2-甲基咪唑的摩尔比为1∶20~100;所述含钴溶液由钴盐溶于甲醇中制备得到;所述钴盐为氯化钴、硝酸钴和硫酸钴中的至少一种;所述2-甲基咪唑溶液由2-甲基咪唑溶于甲醇中制备得到。
[0012]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,步骤s2中,所述酸溶液为硫酸、硝酸、盐酸、单宁酸溶液中的至少一种;所述酸溶液的浓度为0.5mol/l~3.0mol/l;所述浸泡过程中控制体系的温度为80℃~100℃;所述浸泡的时间为8h~24h。
[0013]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述磁性氮掺杂碳的添加量为每升含有机污染物水体中添加磁性氮掺杂碳0.01g~0.2g。
[0014]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述过硫酸盐的添加量为每升含有机污染物水体中添加过硫酸盐0.04mmol~0.40mmol。
[0015]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述过硫酸盐为过一硫酸盐和/或过二硫酸盐;所述过一硫酸盐为过一硫酸钾;所述过二硫酸盐为过硫酸钠、过硫酸钾或过硫酸铵。
[0016]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述含有机污染物水体中的有机污染物为四环素、双酚a或磺胺甲恶唑中的至少一种;所述含有机污染物水体中的有机污染物的浓度为5μmol/l~40μmol/l。
[0017]
上述的利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,进一步改进的,所述降解反应过程中控制反应体系的ph值为3~9;所述降解反应在转速为100rpm~150rpm的搅拌条件下进行;所述降解反应的温度为20℃~50℃;所述降解反应的时间为2min~45min。
[0018]
与现有技术相比,本发明的优点在于:
[0019]
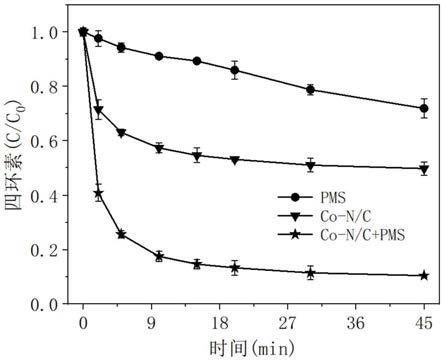
(1)本发明提供了一种利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法,以磁性氮掺杂碳为催化剂,通过利用磁性氮掺杂碳的活化作用对过硫酸盐进行活化,从而产生强活性物质-单线态氧,进而构建得到以单线态氧为主要反应的降解催化体系,最终实现对水体中有机污染物的高效降解。本发明中,采用的磁性氮掺杂碳包括氮掺杂碳,氮掺杂碳中包裹有co纳米颗粒,这是一种具有分层多孔结构、特定氮构型和丰富官能团的新型过硫酸盐活化材料,可以高效活化过硫酸盐产生强活性物质-单线态氧,因而对多种有机
污染物均展现极好的降解性能,且反应时间短,同时还表现出极好的抗水基质干扰能力,几乎不受水中ph、无机阴离子和溶解性有机物的影响,由此可实现对水体中有机污染物的高效去除。更为重要的是,本发明采用的磁性氮掺杂碳中co纳米颗粒被包裹在氮掺杂碳内,在实际催化活化过程中不会有金属co溶出,因而不会造成对水体的二次污染,且包裹在碳基质中的co赋予了催化剂磁性,便于分离和循环使用,有利于降低处理成本。本发明利用磁性氮掺杂碳活化过硫酸盐降解水体中有机污染物的方法具有处理成本低、抗水基质干扰能力强、降解效率高、绿色环保等优点,能够高效去除水体中的有机污染物,使用价值高,应用前景好。
[0020]
(2)本发明中,磁性氮掺杂碳是以zif-67材料为原料经过煅烧和酸刻蚀后制备得到,通过直接热解zif-67材料,可节约试剂成本和减少合成过程,成本更低,工艺更简单。
附图说明
[0021]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整的描述。
[0022]
图1为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的x射线衍射图。
[0023]
图2为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的不同放大倍数扫描电镜图。
[0024]
图3为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的不同放大倍数透射电镜图。
[0025]
图4为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对水体中四环素的降解效果图。
[0026]
图5为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对水体中双酚a和磺胺甲恶唑的降解效果图。
[0027]
图6为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)、纳米零价铁、四氧化三钴、生物炭、碳纳米管、还原氧化石墨烯和钴离子活化过一硫酸盐时对水体中四环素的降解效果图。
[0028]
图7为本发明实施例2中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对不同类型水体中四环素的降解效果图。
[0029]
图8为本发明实施例3中不同处理条件处理后的四环素水体中微藻生长量的柱形图。
具体实施方式
[0030]
以下结合说明书附图和具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
[0031]
以下实施例中所采用的原料和仪器均为市售。以下实施例中,若无特别说明,所得数据均是三次以上重复实验的平均值。
[0032]
实施例1:
[0033]
一种利用磁性氮掺杂碳(co-n/c)活化过硫酸盐降解水体中有机污染物的方法,包括以下步骤:
[0034]
取100ml、摩尔浓度为40.0μm的四环素溶液于250ml锥形瓶中,使用2.0mm硼酸盐缓冲液控制溶液ph在7.0
±
0.1,同时加入磁性氮掺杂碳(co-n/c)和过一硫酸盐(pms),引发反应,其中反应体系中co-n/c和pms的剂量分别为0.1g/l和0.2mmol/l。反应在水浴摇床中进行,转速150rpm,温度30℃,时间45min,完成对水体中四环素的降解。本实施例中,过一硫酸盐(pms)为过一硫酸钾。
[0035]
本实施例中,采用的磁性氮掺杂碳(co-n/c)的制备方法,包括以下步骤:
[0036]
称取2.87克硝酸钴溶解在80毫升甲醇中,得到溶液a(硝酸钴溶液)。称取6.52克2-甲基咪唑溶解在200毫升甲醇中,得到溶液b(2-甲基咪唑溶液)。然后,将溶液a快速倒入溶液b中,在室温下剧烈磁力搅拌,反应24小时后,将产生的紫色产物离心,用甲醇洗涤3次,并在60℃下真空干燥,得到沸石咪唑酯骨架(zif-67材料)。将zif-67材料放入到管式炉中,在氩气气氛下以2℃/min的升温速率到达900℃,煅烧2小时。随后,将黑色产物浸泡在浓度为0.5m的硫酸溶液中,于80℃下搅拌反应12小时,以除去任何不稳定的co类物质。最后,将样品用超纯水洗涤几次,并在60℃真空干燥,得到磁性氮掺杂碳(co-n/c)。
[0037]
将本发明实施例1中制备的磁性氮掺杂碳(co-n/c)用x射线衍射仪、扫描电镜和透射电镜进行结构分析,结果如图1-3所示。
[0038]
图1为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的x射线衍射图。由图1可知,图谱中的特征衍射峰很好地指向原始的zif-67,在900℃碳化和酸刻蚀后,所得的磁性氮掺杂碳(co-n/c)仅显示三个衍射峰,而且,磁性氮掺杂碳(co-n/c)在25.8
°
附近显示一个尖锐的衍射峰,表明石墨化程度得到改善,其他两个在44.2
°
和51.4
°
处的衍射峰对应于表面结构化金属co的(111)和(200)晶面。
[0039]
图2为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的不同放大倍数扫描电镜图。由图2可知,磁性氮掺杂碳(co-n/c)具有清晰的菱形十二面体形状且表面变得粗糙和起皱不平,而且,由于煅烧过程中co的催化作用,一些纳米管出现在催化剂表面,平均直径约为16nm。
[0040]
图3为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)的不同放大倍数透射电镜图。由图3可知,本发明制得的磁性氮掺杂碳(co-n/c)中存在纳米管,该结果与扫描电镜测试的结果一致。同时,在催化剂的外部有石墨烯薄片。另外,通过透射电镜图像观察到co纳米颗粒(约16nm)并嵌入石墨碳层中,这造成很难通过化学蚀刻完全去除。
[0041]
上述这些结果表明,本发明制备的磁性氮掺杂碳(co-n/c)中co纳米颗粒包裹在氮掺杂碳中。
[0042]
本实施例中,在进行液相分析之前,在指定的时间间隔吸取1.0ml样品,用0.45μm过滤器过滤,并加入15μl摩尔浓度为1.0mol/l的硫代硫酸钠溶液淬灭,测定样品中四环素的浓度,进而计算得到四环素的去除率,结果如图4所示。
[0043]
图4为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对水体中四环素的降解效果图。如图4所示结果表明:单独加入pms时可直接缓慢氧化降解四环素,在45分钟内降解效率为28.2%,这对应于pms自身的氧化能力。由于磁性氮掺杂碳(co-n/c)的高比表面积和多孔结构,其可以通过吸附作用去除50.4%的四环素。当两者组合使用时,85.4%的四环素可在15分钟内快速降解,表明磁性氮掺杂碳(co-n/c)能够很好地活化pms以降解四环素。
[0044]
此外,在相同反应条件下,其他有机污染物,如双酚a和磺胺甲恶唑,在30分钟内的降解效率高达100.0%和96.6%(如图5所示)。图5为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对水体中双酚a和磺胺甲恶唑的降解效果图。
[0045]
对比例1:
[0046]
一种利用常规活化剂活化过硫酸盐降解水体中有机污染物的方法,包括以下步骤:
[0047]
取100ml、摩尔浓度为40.0μm的四环素溶液于250ml锥形瓶中,使用2.0mm硼酸盐缓冲液控制溶液ph在7.0
±
0.1,同时加入不同类型的活化剂(包括纳米零价铁、四氧化三钴、生物炭、碳纳米管、还原氧化石墨烯和钴离子)和过一硫酸盐(pms)引发反应,除钴离子浓度设置为5.0mg/l外,其他活化剂的剂量均为0.1g/l,过一硫酸盐的剂量为0.2mmol/l。反应在水浴摇床中进行,转速150rpm,温度30℃,时间45min,完成对水体中四环素的降解。采用的过一硫酸盐(pms)为过一硫酸钾。
[0048]
本实施例中,在进行液相分析之前,在指定的时间间隔吸取1.0ml样品,用0.45μm过滤器过滤,并加入15μl摩尔浓度为1.0mol/l的硫代硫酸钠溶液淬灭,测定样品中四环素的浓度,进而计算得到四环素的去除率,结果如图6所示。
[0049]
图6为本发明实施例1中制得的磁性氮掺杂碳(co-n/c)、纳米零价铁、四氧化三钴、生物炭、碳纳米管、还原氧化石墨烯和钴离子活化过一硫酸盐时对水体中四环素的降解效果图。由图6可知,当活化剂分别为纳米零价铁、四氧化三钴、生物炭、碳纳米管、还原氧化石墨烯和钴离子时,对应的四环素在45分钟内的降解效率为96.0%、42.9%、53.7%、56.0%、58.0%和89.6%,这表明本发明制备的磁性氮掺杂碳(co-n/c)的催化活性不仅优于常用的pms活化剂(co3o4,碳纳米管,还原氧化石墨烯和生物炭),而且可与最先进的催化剂(nzvi和co
2+
)媲美。同时,采用本发明磁性氮掺杂碳(co-n/c)的催化过程中,钴溶出浓度基本无法用电感耦合电离子发射光谱检出,因而磁性氮掺杂碳(co-n/c)具有更好的应用前景。
[0050]
实施例2
[0051]
考察本发明磁性氮掺杂碳(co-n/c)对不同类型水样的适用性,包括以下步骤:
[0052]
分别从湘江长沙段和桃子湖(中国长沙)采集水样以及在实验室采集自来水样,向其中喷射四环素储备液以获得初始浓度为40.0μm的四环素溶液,从而模拟实际水样。取100ml上述三种实际水样于250ml锥形瓶中,同时加入实施例1中制得的磁性氮掺杂碳(co-n/c)和过一硫酸盐(pms)引发反应,其中体系中co-n/c和pms的剂量分别为0.1g/l和0.2mmol/l。反应在水浴摇床中进行,转速150rpm,温度30℃,时间为45min,完成对水体中四环素的降解。采用的过一硫酸盐(pms)为过一硫酸钾。
[0053]
在进行液相分析之前,在指定的时间间隔吸取1.0ml样品,用0.45μm过滤器过滤,并加入15μl摩尔浓度为1.0mol/l的硫代硫酸钠溶液淬灭,测定样品中四环素的浓度,进而计算得到四环素的去除率,结果如图7所示。
[0054]
图7为本发明实施例2中制得的磁性氮掺杂碳(co-n/c)活化过一硫酸盐时对不同类型水体中四环素的降解效果图。由图7可知,构建的催化体系在实际水样中可高效去除水中四环素并受实际水体的影响很小,这表明co-n/c-pms体系在实际水体中具有很高的适应性,因而具有很好的应用前景。
[0055]
实施例3
[0056]
取100ml、摩尔浓度为40.0μm的四环素溶液于250ml锥形瓶中,使用2.0mm硼酸盐缓冲液控制溶液ph在7.0
±
0.1,同时加入实施例1中制得的磁性氮掺杂碳(co-n/c)和过一硫酸盐(pms),引发反应,其中反应体系中co-n/c和pms的剂量分别为0.1g/l和0.2mmol/l。反应在水浴摇床中进行,转速150rpm,温度30℃,时间45min,完成对水体中四环素的降解。采用的过一硫酸盐(pms)为过一硫酸钾。
[0057]
反应45分钟后,取其溶液(单独pms、co-n/c吸附以及co-n/c和pms联合处理)以及空白水样和未处理的原始四环素溶液,总共五组样品。过滤后,取45ml滤液加到100ml锥形瓶中。然后,加入5ml微藻coelastrella sp.浓缩悬浮液并混匀,将其置于恒温培养箱中进行光照射培养。光照条件为每天光照8小时,黑暗条件维持16小时。定期取样并用紫外可见光度计在680nm吸光度下测定微藻的生长量。样品初始吸光度为0.134。微藻coelastrella sp.的干重计算如式1所示。
[0058]
dw(mg/l)=0.3357
×
od
680
×
1000(r2=0.9962)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(1)
[0059]
图8为本发明实施例3中不同处理条件处理后的四环素水体中微藻生长量的柱形图。由图8可知,相比于对照组,微藻coelastrella sp.的生长量在所有体系中均被抑制;其中在未处理的四环素溶液中,抑制作用随培养时间的延长变得更加显著,在第六天其抑制率高达53.7%。当通过co-n/c或单独pms处理四环素溶液时,其抑制作用没有明显缓解。相反,在通过co-n/c-pms体系联合处理后,微藻coelastrella sp.的生长从第三天开始加速,到第六天抑制率降低至29.9%,表明毒性降低。同时,该体系也优于以往的pms类高级氧化技术去除四环素,处理后其毒性反而增加。
[0060]
由此上述结果可知,本发明中利用磁性氮掺杂碳构建的催化体系可高效降解水中有机污染物,其性能优于大多数报道的pms活化剂,甚至最先进的pms活化剂(纳米零价铁和钴离子),耐受水基质的干扰,在实际模拟水样中展现高效能,特别是经过联合处理后四环素的生态毒性显著降低,这些优势为水中有机污染物的去除提供了一种高效方法。
[0061]
以上实施例仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例。凡属于本发明思路下的技术方案均属于本发明的保护范围。应该指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下的改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1