一种减少副产物碳层的碳纳米线圈合成用催化剂的制备方法及其应用与流程
本发明属于材料制备技术领域,涉及一种减少副产物碳层碳纳米线圈合成用催化剂的制备方法及其应用。
背景技术:
碳纳米线圈由于其独特的三维螺旋纳米结构,使其具备优异的电磁特性、机械特性及分散特性,可以广泛应用于电磁吸波材料、应变传感器件、微纳米机电系统、三维纳米螺旋模板等场合。目前,已经公开的制备碳纳米线圈的制备方法有很多,其中最适合应用于工业生产的是化学气相沉积法(cvd法),该方法将催化剂担载在衬底表面,置于化学气相沉积系统内,使碳源气体在一定反应条件下流经催化剂表面发生催化分解沉积。使用该方法合成碳纳米线圈的过程中,选择高效的催化剂十分关键,催化剂的形貌、组分及颗粒大小都会影响碳纳米线圈的合成质量。
目前公开的适合碳纳米线圈生长的催化剂主要包括铁、钴、镍、钨过渡金属及其合金,尤其是以铁锡氧化物合金(fe-sn-o)颗粒为代表的催化剂被广泛证明可以高效低成本合成碳纳米线圈。目前公开的制备fe-sn-o催化剂的方法主要包括溶液法(出版物:d.w.lietal.,advancedmaterialsresearch,vols.60-61,pp.251-,255,2009)、溶胶凝胶法(出版物:d.w.lietal.,carbon,48(1):170-175,2010.)、离子溅射法(出版物:fu,xin,etal.,carbon99(2016):43-48.,2016.),以上三种方法都可以成功制备用于合成碳纳米线圈的fe-sn-o催化剂,但是它们共同存在的问题就是在合成碳纳米线圈的过程中,会在合成的碳纳米线圈底部生长出数微米到数十微米厚的非晶碳层,使得合成的碳纳米线圈纯度大大降低,严重限制其大规模应用;造成这种现象的主要原因是使用的催化剂中并非全部都适合合成碳纳米线圈,一部分催化剂只能合成非晶碳层,使得制备的碳纳米线圈的整体纯度显著下降,并且额外引入提纯的问题。因此制备用于合成无副产物碳层碳纳米线圈的高效均一催化剂受到越来越多科研人员的关注。
技术实现要素:
本发明的目的是针对现有技术中碳纳米线圈合成过程中存在副产物非晶碳层,碳纳米线圈纯度不高的问题,提供一种减少副产物碳层碳纳米线圈合成用催化剂、制备方法及其应用。
为实现上述目的,本发明所采用的技术方案是:
一种减少副产物碳层的碳纳米线圈合成用催化剂的制备方法,包括以下步骤:
(a)以可溶性fe3+或fe2+盐和可溶性sn4+或sn2+盐为原料,按fe:sn原子摩尔比60:1~3:1的比例溶解在还原性溶剂中,混合均匀后得到催化剂前驱体溶液,催化剂前驱体溶液中fe3+或fe2+的浓度范围为0.01-0.05mol/l。
(b)将步骤(a)制备的催化剂前驱体溶液转移至高温反应釜,在溶剂热体系中控制反应温度在160-220℃,在高压釜溶剂热反应5-30小时后,自然冷却到室温,将所得的红色沉淀过滤、洗涤、干燥,得到红色单一的fe-sn-o催化剂粉末,粒径分布在100nm-500nm之间。
所述的fe:sn原子摩尔比优选为10:1。
所述的还原性溶剂包括n,n-二甲基甲酰胺(dmf)、乙腈、乙二醇、异丙醇等,优选为dmf。
所述的可溶性fe3+或fe2+盐包括但不限于硝酸(亚)铁、氯化(亚)铁、-、硫酸铁,可溶性sn4+或sn2+盐包括但不限于氧化(亚)锡、氯化(亚)锡,且fe3+或fe2+盐与sn4+或sn2+盐可以任意组合。
一种减少副产物碳层的碳纳米线圈合成用催化剂的应用,采用该催化剂合成少甚至无副产物碳层的碳纳米线圈,包括以下步骤:
室温下,将制备得到的催化剂粉末分散至水或有机溶液中得到催化剂分散液,其中,催化剂浓度为0.5-5mg/ml;将催化剂分散液附着至衬底表面后干燥,或将催化剂粉末直接附着于或通过表面修饰附着于衬底表面。将担载催化剂的衬底至于cvd系统中,以乙炔为碳源,氩气、氮气或其他惰性气体为保护气,在650℃-800℃的反应温度下反应3min-300min;生长得到少甚至无副产物碳层的碳纳米线圈。
所述的衬底包括石英、硅、sio2、石墨(烯)、不锈钢、金属氧化物、硼化物、氮化物、纤维、陶瓷等包括平板和球体在内的各种形状的固体衬底。
所述的cvd系统中,碳源流速为15sccm,保护气流量为245sccm。
与现有技术相比,本发明的有益效果为:本发明通过一步溶剂热法制备用于合成少甚至无副产物碳层的碳纳米线圈的催化剂,制备方法简单,易于操作,工艺简单,成本低廉;由本发明制备得到的催化剂合成的碳纳米线圈,具有纯度高、少甚至无副产物碳层的特点,有益于大规模工业制备,具有突出的应用前景。
附图说明
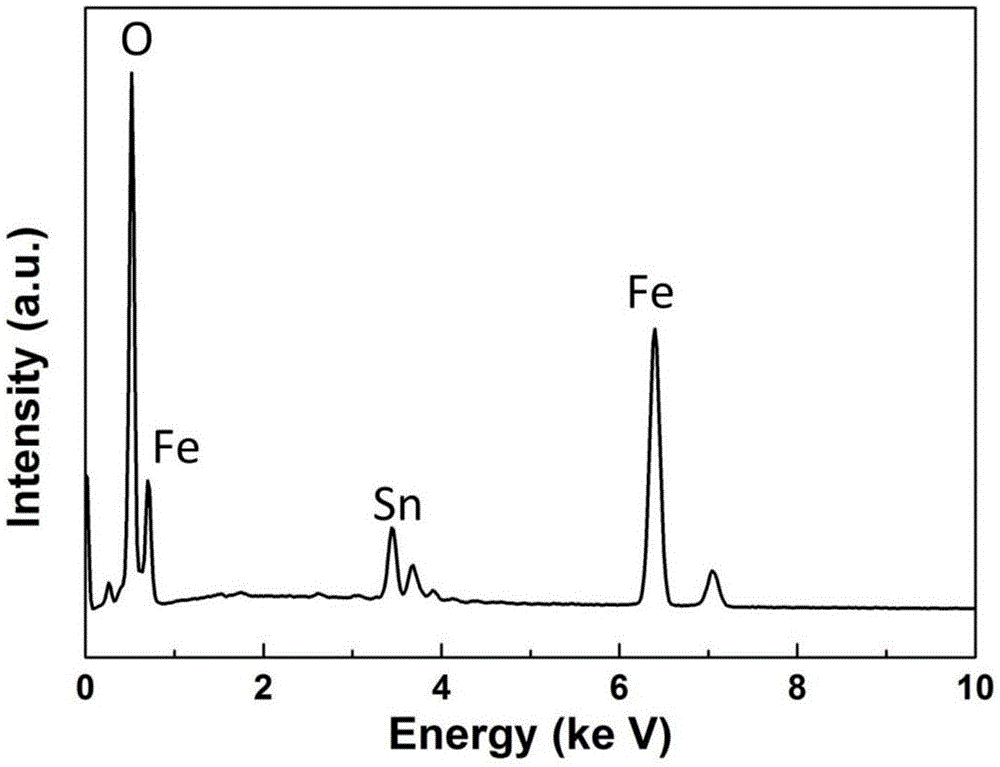
图1是实施例1制备的催化剂粉末的eds元素分析测试图谱。
图2是实施例1制备的催化剂粉末的x射线衍射图(xrd)。
图3是实施例1制备的催化剂粉末的x射线光电子能谱分析图(sn元素部分)。
图4是实施例1制备催化剂粉末的扫描电镜图(sem)。
图5是实施例1制备的碳纳米线圈俯视扫描电镜图(sem)。
图6是实施例1制备的碳纳米线圈的截断面扫描电镜图(sem)。
图7是实施例2制备的催化剂粉末的eds元素分析测试图谱。
图8是实施例2制备的碳纳米线圈的截断面扫描电镜图(sem)。
图9是实施例3制备的碳纳米线圈的截断面扫描电镜图(sem)。
图10.是实施例4制备的碳纳米线圈的截断面扫描电镜图(sem)。
具体实施方式
以下结合附图对本发明最进一步说明。
本发明实施方案的用于制备减少产物副产物碳层的碳纳米线圈的催化剂主要由氧化铁(α-fe2o3)和氧化锡(sno2)聚集体颗粒组成。
实施例1:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂
将404mgfe(no3)3·9h2o和35.06mgsncl4·5h2o(fe:sn原子摩尔比为10:1)溶于36mln,n-二甲基甲酰胺(dmf),超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在180℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
附图1为催化剂粉末的eds元素分析测试结果,测试结果表明,红色粉末主要由fe、sn、o三种元素组成;附图2为该催化剂粉末的x射线衍射图(xrd),可以看出该图谱与α-fe2o3的标准图谱pdf#33-0664峰位一致,表明催化剂中的fe元素是来自α-fe2o3,由于产物中sn元素的氧化物结晶性不好,x射线衍射图不能确定其具体价态,需要进一步测试确认。附图3为催化剂粉末的x射线光电子能谱分析图(xps),图中可以看到sn3d5/2和sn3d3/2结合能的峰出现在486.2ev和494.6ev处,根据xps标准结合能对照表可以确定与sno2中sn元素+4价氧化态相一致,因此确定催化剂中sn元素来自sno2。附图4是制备催化剂粉末的扫描电镜图(sem),图中可见该粉末为疏松多孔的聚集体结构,利于下一步反应过程中催化剂对碳源气体的吸附,催化剂颗粒分布范围100nm-500nm。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取步骤(1)制备的催化剂粉末分散至酒精中,浓度0.5mg/ml,取反应担载衬底硅片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液滴涂至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为245sccm,反应温度为700℃,反应时间为30min,待反应结束后自然降温。
附图5为反应后的典型产物俯视扫描电镜图(sem),可以看到大量的碳纳米线圈被合成,纯度高于95%。附图6为反应典型产物的截断面扫描电镜图,从图中可以看到,产物底部与衬底接触面多为碳纳米线圈和碳纳米纤维,无致密厚实的非晶碳层产生,使得反应产物中碳纳米线圈的纯度大幅度提高。
实施例2:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将273mgfecl3·6h2o和35.06mgsncl4·5h2o(fe:sn原子摩尔比为10:1)加入36ml乙腈,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在220℃,反应时间为5小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
附图7是实施例2制备的催化剂粉末的eds元素分析测试图谱,测试结果表明,红色粉末主要由fe、sn、o三种元素组成,同时存在si的峰是来自衬底硅片。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至酒精中超声待用,浓度为1mg/ml,取反应担载衬底石英片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为245sccm,反应温度为650℃,反应时间为120min。待反应结束后自然降温。附图8为反应典型产物的截断面扫描电镜图,从图中可以看到,产物底部与衬底接触面多为碳纳米线圈和碳纳米纤维,无致密厚实的非晶碳层产生。
实施例3:
(1)用于制备减少产物副碳层的碳纳米线圈的催化剂。
将200mgfe2(so4)3·9h2o和35.06mgsncl2·2h2o(fe:sn原子摩尔比为10:1)溶于36ml异丙醇,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在160℃,反应时间为20小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用,浓度1mg/ml,取反应担载衬底石墨基板分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底石英片表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为710℃,反应时间为90min。待反应结束后自然降温。
附图9为反应典型产物的截断面扫描电镜图,从图中可以看到,产物底部与衬底接触面多为碳纳米线圈和碳纳米纤维,无致密厚实的非晶碳层产生。
实施例4:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将287mgfe(no3)2·6h2o和35.06mgsncl4·5h2o(fe:sn原子摩尔比为10:1)溶于36ml乙二醇中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在200℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用,浓度为:5mg/ml,取反应担载不锈钢衬底分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氮气(n2)为保护气,流量为240sccm,反应温度为710℃,反应时间为30min。待反应结束后自然降温。附图10为反应典型产物的截断面扫描电镜图,从图中可以看到,产物底部与衬底接触面多为碳纳米线圈和碳纳米纤维,无致密厚实的非晶碳层产生。
实施例5:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将404mgfe(no3)3·9h2o和35.06mgsncl4·5h2o(fe:sn原子摩尔比为10:1)溶于36ml乙二醇中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在200℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用(浓度为:1mg/ml),取反应担载衬底硅片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氮气(n2)为保护气,流量为240sccm,反应温度为710℃,反应时间为30min。待反应结束后自然降温。产物即为少甚至无副产物碳层的碳纳米线圈。
实施例6:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将198mgfecl2·4h2o和35.06mgsncl4·5h2o(fe:sn原子摩尔比为10:1)溶于36ml异丙醇中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在220℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用,浓度3mg/ml,取反应担载衬底陶瓷基片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为710℃,反应时间为30min。待反应结束后自然降温。产物即为无副产物碳层的碳纳米线圈。
实施例7:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将404mgfe(no3)3·9h2o和105.18mgsncl4·5h2o(fe:sn原子摩尔比为3:1)溶于100mln,n-二甲基甲酰胺(dmf)中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在180℃,反应时间为5小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用(浓度为:5mg/ml),取反应担载衬底sio2分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为650℃,反应时间为120min。待反应结束后自然降温。产物即为少甚至无副产物碳层的碳纳米线圈。
实施例8:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将404mgfe(no3)3·9h2o和5.26mgsncl4·5h2o(fe:sn原子摩尔比为60:1)溶于10mln,n-二甲基甲酰胺(dmf)中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在180℃,反应时间为15小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用(浓度为:2mg/ml),取反应担载衬底硅片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底石墨基板表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为800℃,反应时间为10min。待反应结束后自然降温。产物即为少甚至无副产物碳层的碳纳米线圈。
实施例9:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将278mgfeso4·7h2o和22.56mgsncl2·2h2o(fe:sn原子摩尔比为10:1)溶于36mln,n-二甲基甲酰胺(dmf)中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在180℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用(浓度为:2mg/ml),取反应担载衬底氧化铝小球分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底石墨基板表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为800℃,反应时间为10min。待反应结束后自然降温。产物即为少甚至无副产物碳层的碳纳米线圈。
实施例10:
(1)用于制备减少副产物碳层的碳纳米线圈的催化剂。
将287mgfe(no3)2·6h2o和22.56mgsncl2·2h2o(fe:sn原子摩尔比为10:1)溶于36ml乙腈中,超声至混合溶液完全溶解,将混合分散均匀后的混合溶液转移至高温高压反应釜内,在溶剂热体系中控制反应温度在180℃,反应时间为30小时,自然冷却到室温,将所得的红色沉淀过滤,洗涤,干燥,得到单一红色粉末。
(2)使用上述催化剂制备少甚至无副产物碳层的碳纳米线圈
准确称取一定量步骤(1)制备的催化剂粉末分散至水或有机溶液中超声待用(浓度为:2mg/ml),取反应担载衬底硅片分别用丙酮、酒精、去离子水清洗后干燥待用。量取1ml催化剂分散液涂布至衬底石墨基板表面;待干燥后将担载催化剂的衬底至于cvd系统中,以乙炔(c2h2)为碳源,流速为15sccm,氩气(ar)为保护气,流量为240sccm,反应温度为800℃,反应时间为10min。待反应结束后自然降温。产物即为少甚至无副产物碳层的碳纳米线圈。
上述实例证明:采用本文提出的技术方案设计合成的fe-sn-o催化剂颗粒可以使用简便的cvd方法高效制备少甚至无副产物非晶碳层的碳纳米线圈,显著提高碳纳米线圈合成的纯度。
同时上述对实例的描述是为便于该技术领域的普通技术人员能理解和应用本发明。所述实施例仅表达了本发明的实施方式,但并不能因此而理解为对本发明专利的范围的限制,以上应当指出,对于本领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!