一种聚结型对位二取代基苯吸附剂及其制备方法与流程
1.本发明涉及一种聚结型分子筛吸附剂及其制备方法,具体地说,是一种吸附分离对位二取代基苯的吸附剂及其制备方法。
背景技术:
2.对二甲苯是重要的基础化工原料,主要用于生产聚酯纤维。目前,工业上普遍采用吸附分离方法从混合c8芳烃中分离对二甲苯。吸附分离技术包括可选择性吸附对二甲苯的吸附剂和连续逆流的模拟移动床吸附分离工艺。其中,高性能吸附剂的制备是获得高纯度对二甲苯产品的关键。
3.工业用对二甲苯吸附分离吸附剂的活性组元多为x分子筛,将x分子筛与粘土按一定比例混合均匀,经滚球成型、干燥、焙烧和阳离子交换后得到吸附剂小球。抗压强度、选择性、吸附容量和传质性能是评价吸附剂的重要指标。
4.us3558730公开了一种bakx分子筛,其对碳八芳烃中对二甲苯的选择性明显高于bax和kx。
5.cn1275926a公开了一种聚结型沸石吸附剂,活性组元为si/al原子比为 1~1.15的x分子筛,粘结剂为可沸石化的粘土。经碱处理后可使粘土转化为x 分子筛,获得较高的抗压强度和吸附容量。
6.cn1565718a公开了一种对二甲苯吸附剂及制备方法,采用晶粒粒径为 0.1~0.4微米的小晶粒x分子筛作为吸附剂的活性组元,以提高吸附剂的传质性能和提高吸附容量。
7.cn101497022a公开了一种聚结型吸附剂及其制备方法,该法通过在制备吸附剂的混合粉料中加入造孔剂,使转晶后的吸附剂颗粒内形成大量孔径为 100~500纳米的晶间孔,从而显著提高吸附剂的传质性能。
技术实现要素:
8.本发明的目的是提供一种聚结型对位二取代基苯的吸附剂及其制备方法,该吸附剂具有较高的对位二取代基苯的吸附选择性和吸附容量。
9.本发明提供的聚结型对位二取代基苯吸附剂,包括96.5~99.8质量%的x 分子筛和0.2~3.5质量%的基质,所述x分子筛由非转晶x分子筛和转晶生成的x分子筛组成,所述非转晶x分子筛的sio2/al2o3摩尔比为2.1~2.6,其骨架结构中,si(osi)(oal)3结构四面体的含量为30~60mol%,且没有si(osi)4结构的四面体,所述x分子筛的阳离子位为ⅱa族金属或者为ⅱa族金属和
ⅰꢀ
a族金属共同占据。
10.本发明吸附剂活性组分x分子筛中的非转晶x分子筛的骨架结构中含有较多的si(osi)(oal)3结构的四面体,用于从二取代基苯化合物中吸附分离对位二取代基苯异构体,具有较高的吸附选择性,并可提高分离产品纯度,从而提高吸附分离装置的生产能力。
附图说明
11.图1为本发明实例1制备的x分子筛的xrd谱图。
12.图2为本发明实例1制备的x分子筛的
29
si固体核磁共振谱图。
13.图3为对比例1制备的x分子筛的
29
si固体核磁共振谱图。
14.图4为小型模拟移动床吸附分离示意图。
具体实施方式
15.x分子筛的骨架结构中si与si、si与al通过si-o-si、si-o-al相连,形成的四面体中含有五种不同的结构,可用通式si(osi)
4-n
(oal)
n
表示,通式中的 n=0、1、2、3、4,其中含有
“-
o-al”键的四面体中,由于铝和硅的电性不同,导致分子筛具有吸附电性。通常,si-o-al的数量随着分子筛骨架硅/铝比的降低而升高。然而,在分子筛骨架硅/铝比基本相等的前提下,不同合成方法制备的x分子筛骨架结构中,si(osi)
4-n
(oal)
n
所示的各种四面体的含量存在差异。
16.本发明将具有特定骨架结构的x分子筛(非转晶x分子筛)与作为粘结剂的高岭土矿物混合后滚球成型,在高温下焙烧,使其中的高岭土矿物转化为偏高岭土,然后再通过碱处理,使偏高岭土原位晶化转化成x分子筛。所述的具有特定骨架结构的x分子筛在合成过程中加入能和al形成配合物的多元羧酸根离子,其骨架中可形成较多的si(osi)(oal)3结构四面体,并且不含n=0的硅氧四面体结构,从而提高了吸附剂对位二取代基苯的吸附选择性。
17.本发明所述吸附剂中含两种x分子筛,一种是非转晶x分子筛,具有特定的骨架结构,另一种是吸附剂成型过程中使用的粘结剂,一般为高岭土矿物原位晶化后形成的x分子筛。所述吸附剂优选包括97.5~99.8质量%的x分子筛和0.2~2.5质量%的基质。
18.所述非转晶x分子筛的骨架结构中,si(osi)(oal)3结构四面体的含量优选 40~50mol%。
19.所述吸附剂中非转晶x分子筛的晶粒粒径优选0.5~1.6微米。
20.本发明吸附剂中x分子筛的阳离子位为ⅱa族金属或者为ⅱa族金属和ⅰa族金属共同占据。所述的ⅱa族金属优选ba,ⅰa族金属优选k、li和na 中的至少一种。当x分子筛的阳离子位由ba离子和ⅰa族金属离子共同占据时,其中氧化钡与ⅰa族金属氧化物的摩尔比为1~50、优选1~40。
21.吸附剂中所述的基质为高岭土矿物经原位晶化转晶后的剩余物。所述的高岭土矿物选自高岭石、地开石、珍珠石、耐火石和埃洛石中的至少一种。
22.本发明所述的吸附剂优选为小球状,小球的平均粒径优选300~850微米。
23.本发明所述吸附剂的制备方法,包括:
24.(1)将非转晶x分子筛与高岭土矿物按88~95:5~12的质量比混合均匀,滚球成型制成小球,干燥后于500~700℃焙烧;
25.(2)将(1)步焙烧后所得小球用氢氧化钠与氢氧化钾的混合溶液处理,使其中的高岭土矿物原位晶化为x分子筛,然后干燥;
26.(3)用ⅱa族金属的可溶性盐溶液或者是ⅱa族金属和ⅰa族金属的可溶性盐的混合溶液对(2)步干燥后的小球进行阳离子交换,干燥、活化。
27.本发明方法(1)步是将非转晶x分子筛与高岭土矿物滚球成型,所述的高岭土矿物选自高岭石、地开石、珍珠石、耐火石、埃洛石或它们的混合物。所述高岭土矿物中晶化物质的含量至少为90质量%、优选93~99质量%。
28.(1)步滚球成型的设备可为转盘、糖衣锅或滚筒。滚球成型时,将混合均匀的固体原料放入转动设备中,边滚动边喷水使固体粉末粘附团聚成小球。滚球时水的加入量为固体总质量的6~22%,优选6~16%。
29.(1)步滚动成球后的小球,经过筛分,取一定范围粒径的小球,将其干燥、焙烧制得吸附剂。所述干燥温度优选60~110℃,时间优选2~12小时,焙烧温度优选530~700℃,时间优选1.0~6.0小时。经过焙烧后,小球内的高岭石转化为偏高岭土,以便于(2)步转晶为x分子筛。
30.本发明方法(2)步为将(1)步制备的小球进行原位晶化,原位晶化所用的碱液为氢氧化钠和氢氧化钾混合溶液,其中氢氧根离子的浓度为 0.1~3.0mol/l、优选0.2~1.6mol/l,k/(na+k)摩尔比为0.1~0.6、优选0.15~0.45。
31.(2)步中用氢氧化钠与氢氧化钾的混合溶液对高岭土矿物进行原位晶化处理的液/固比优选1.5~5.0l/kg。所述高岭土矿物原位晶化为x型分子筛的晶化温度优选80~100℃、更优选85~100℃,时间优选0.5~8小时。将原位晶化后的小球干燥,所述的干燥温度优选60~110℃,时间优选2~12小时。
32.本发明方法中(3)步为对(2)步干燥后的小球进行阳离子交换,所述
ⅱꢀ
a族金属的可溶性盐优选硝酸钡或氯化钡,ⅰa族金属的可溶性盐优选其硝酸盐和氯化物中的一种。阳离子交换可在釜式或柱式容器中进行,优选在柱式容器中连续交换。进行离子交换的温度为60~160℃,优选80~110℃。交换液中的阳离子摩尔数与沸石中钠离子摩尔数之比,即交换比优选1.5~3.0。若吸附剂中同时含有钡和钾时,可配制钡盐和钾盐的混合溶液作为交换液同时进行钡、钾离子交换,也可先用钡盐溶液进行钡交换,再用钾盐溶液进行钾交换。所述干燥、活化可在流动的热空气或氮气中进行,所述干燥温度优选40~120℃、更优选60~110℃,时间优选5~60小时、更优选18~40小时。所述的活化温度优选150~250℃、更优选160~220℃,时间优选5~20小时、更优选5~10小时。
33.本发明所述非转晶x分子筛的制备方法,包括如下步骤:
34.(
ⅰ
)将硅源、铝源、水和氢氧化钠混合,各物料摩尔比为 sio2/al2o3=10~25,na2o/sio2=0.6~1.8,h2o/sio2=10~50,在0~60℃老化1~72 小时,制成导向剂,
35.(
ⅱ
)将氢氧化钠、铝源、硅源、(
ⅰ
)步制备的导向剂、多元羧酸钠和水混合均匀形成分子筛合成体系,其中各物料的摩尔比为:sio2/al2o3=2.5~3.2, na2o/sio2=1.5~2.5,h2o/sio2=50~100,多元羧酸钠/al2o3=0.03~0.30,所加导向剂中所含al2o3与分子筛合成体系中所含al2o3的摩尔比为0.01~1.0%,
36.(
ⅲ
)将(
ⅱ
)步中的分子筛合成体系于90~150℃水热晶化2~48小时,晶化后所得固体经洗涤、干燥,得到x分子筛。
37.上述方法所述的铝源优选低碱度偏铝酸钠溶液、氧化铝、氢氧化铝、硫酸铝、氯化铝、硝酸铝和铝酸钠中的一种或几种,优选低碱度偏铝酸钠。所述低碱度偏铝酸钠溶液中al2o3含量为7~15质量%,na2o含量为7~20质量%。
38.所述的硅源选自正硅酸乙酯、硅溶胶、水玻璃、硅酸钠、硅胶和白炭黑中的一种或
几种,优选硅溶胶或水玻璃。
39.上述方法(
ⅰ
)步为合成导向剂,各物料摩尔比优选为sio2/al2o3=12~20, na2o/sio2=0.6~1.5,h2o/sio2=10~30,老化温度优选10~45℃,时间优选10~30 小时。
40.上述方法(
ⅱ
)步为制备分子筛合成体系,所述合成体系中,各物料的摩尔比优选为:sio2/al2o3=2.6~3.1,na2o/sio2=1.5~2.0,h2o/sio2=60~90,多元羧酸钠/al2o3=0.03~0.3,所加导向剂中所含al2o3与分子筛合成体系中所含 al2o3的摩尔比优选为0.05~0.5%。所述的多元羧酸钠的碳原子数为2~5,含有的羧酸根为2~3个,或者上述多元羧酸钠的碳原子数为3~5,含有的羧酸根为 2~3个,并且含有羟基。所述的不带羟基的多元羧酸钠可为草酸钠,带羟基的多元羧酸钠可为柠檬酸钠、酒石酸钠或苹果酸钠。所述的多元羧酸钠优选为草酸钠、柠檬酸钠、酒石酸钠和苹果酸钠中的一种或几种。
41.上述方法(
ⅲ
)步将分子筛合成体系进行水热晶化制备分子筛,所述的水热晶化温度优选90~130℃,时间优选5~20小时、更优选8~15小时。晶化后所得固体经洗涤后的干燥温度优选70~100℃,时间优选2~20小时。
42.本发明提供的吸附剂适用于从混合二取代基苯化合物中吸附分离对位二取代基苯异构体,如从混合碳八芳烃中吸附分离对二甲苯,也可用于从二乙基苯中吸附分离对二乙苯或从甲酚中吸附分离对位异构体。所述二取代基苯的取代基优选c1~c2的烷基或羟基。
43.优选地,采用液相吸附分离过程从混合碳八芳烃中吸附分离对二甲苯。所述液相吸附分离可采用多柱串联方式,也可采用借助旋转阀或电磁阀组实现的模拟移动床进行。吸附分离的操作压力为0.3~1.5mpa,操作温度为120~180℃。吸附分离所使用的解吸剂可以是对二乙苯或甲苯。
44.吸附剂的吸附选择性和对吸附目的组分的吸附、解吸速率是评价吸附剂性能的重要指标。选择性为吸附平衡时,吸附相中两组分浓度的比值与非吸附相中该两组分浓度的比值之比。所述吸附平衡是指混合c8芳烃(也可为其它可分离的二取代基苯混合物)与吸附剂接触后,吸附相和非吸附相之间不发生组分净转移时的状态。吸附选择性的计算公式如下:
[0045][0046]
其中,c和d表示欲进行分离的两种组分,a
c
和a
d
分别表示吸附平衡时吸附相中c、d两种组分的浓度,u
c
和u
d
分别表示吸附平衡时非吸附相中c、 d两种组分的浓度。当两种组分的选择性β≈1.0时,表明吸附剂对两种组分的吸附能力相当,不存在被优先吸附的组分。当β大于或小于1.0时,表明一种组分被优先吸附。具体地说,当β>1.0时,吸附剂优先吸附c组分;当β<1.0 时,吸附剂优先吸附d组分。从分离的难易程度讲,β值越大,吸附分离越容易进行。较快的吸附、解吸速率,有利于减少吸附剂和解吸剂的用量,提高产品收率,降低吸附分离装置的操作费用。
[0047]
本发明使用一种动态脉冲实验装置测定吸附选择性和对二甲苯的吸附、解吸速率。该装置由进料系统、吸附柱、加热炉、压力控制阀等组成。吸附柱为ф6
×
1800毫米的不锈钢管,吸附剂装量为50毫升。吸附柱下端入口与进料和氮气系统相连,上端出口接压力控制阀,再与流出物收集器连接。实验所用解吸剂为30体积%的对二乙苯(pdeb)和70体积%的正庚烷,也可以是30体积%的甲苯(t)和70体积%的正庚烷。脉冲液组成为各占5体积%
的乙苯(eb)、对二甲苯(px)、间二甲苯(mx)、邻二甲苯(ox)、正壬烷(nc9)和75 体积%的所述含正庚烷的解吸剂。
[0048]
选择性的测定方法为:将称量好的吸附剂装入吸附柱震实,在氮气气氛中于160~190℃脱水活化;再通入解吸剂排除系统中的气体;然后将系统压力升至0.8mpa,温度升至177℃(对二乙苯为解吸剂)或者135℃(甲苯为解吸剂),停止通入解吸剂,以1.0小时-1
的体积空速通入8毫升脉冲液,之后切换并以同样的体积空速通入解吸剂,每隔2分钟取3滴脱附液样品,用气相色谱分析。以脱附用解吸剂体积为横坐标,nc9、eb、px、mx和ox各组分浓度为纵坐标,绘制出上述各组分的脱附曲线。其中,nc9不被吸附,可作为示踪剂来获得吸附系统的死体积。将示踪剂半峰宽的中点作为零点,测定eb、px、mx、 ox各组分半峰宽中点到零点的净保留体积,任意组分的净保留体积与吸附平衡时的分配系数成正比,反映了各组分与吸附剂间的作用力,两组分净保留体积之比即为选择性β。
[0049]
为了表示px的吸附、解吸速率和px与pdeb或者t之间的吸附选择性,引入px的吸附速率[s
a
]
10-90
和解吸速率[s
d
]
90-10
。吸附速率[s
a
]
10-90
为px的脉冲脱附曲线中px浓度从10%上升到90%所需的解吸剂体积,解吸速率[s
d
]
90-10
为脉冲脱附曲线中px浓度从90%下降到10%所需的解吸剂体积。[s
a
]
10-90
和 [s
d
]
90-10
的值越小,px的吸附和解吸速率越快。二者比值[s
a
]
10-90
/[s
d
]
90-10
定义为px与解吸剂之间的吸附选择性β
px/pdeb
或β
px/t
。β
px/pdeb
或β
px/t
远小于1.0 表示吸附剂对解吸剂的选择性更强,这对吸附分离过程是不利的,理想的状况β
px/pdeb
或β
px/t
是约等于或稍大于1.0。
[0050]
下面通过实例进一步说明本发明,但本发明并不限于此。
[0051]
实例中,采用甲苯气相吸附实验测定吸附剂的吸附容量,具体操作方法为:在35℃下,使携带甲苯的氮气(甲苯分压为0.05mpa)与一定质量的吸附剂接触,直到甲苯达到吸附平衡。根据甲苯吸附前后吸附剂的质量差由下式计算出被测吸附剂的吸附容量。
[0052][0053]
其中,c为吸附容量,单位为毫克/克;m1为吸附甲苯前被测吸附剂的质量,单位为克;m2为吸附甲苯后被测吸附剂的质量,单位为克。
[0054]
吸附剂抗压强度测定方法:采用dl-ii型颗粒强度测定仪(大连化工研究设计院生产)测定,吸附剂小球过300微米的筛后,在不锈钢筒体中装入约1.5 毫升吸附剂。测定时安装一个与不锈钢筒体过盈配合的顶针,在预先设定好的压力下压一次后倒出吸附剂,再过300微米的筛称重,吸附剂加压测试前后质量减少量为在设定压力下吸附剂的破碎率。
[0055]
吸附剂灼基堆密度的测定方法:在100ml量筒中加入50ml吸附剂,在振实密度仪(辽宁仪表研究所有限责任公司生产)上振动5分钟,再加入50ml 吸附剂并振动5分钟,量筒中吸附剂质量与体积之比为吸附剂堆密度;取一定质量的吸附剂于600℃灼烧2小时,并置于干燥器中冷却至室温,灼烧后与灼烧前吸附剂质量之比为灼基,灼基与吸附剂堆密度之积为灼基堆密度。
[0056]
实例1
[0057]
(1)制备铝源
[0058]
将200kg氢氧化铝、232.15kg氢氧化钠和652.33kg去离子水加入反应釜中,加热至100℃,搅拌6小时,形成澄清透明的低碱度偏铝酸钠溶液,作为铝源。所述铝源中al2o3含量
为11.87质量%,na2o含量为16.59质量%,na2o与al2o3的摩尔比为2.3。
[0059]
(2)制备导向剂
[0060]
在搅拌条件下,将3.81kg氢氧化钠、8.86kg去离子水、4.48kg(1)步制备的铝源和23.24kg的水玻璃(水玻璃中sio2含量为20.17质量%,na2o含量为6.32质量%,下同)加入反应釜中,其中各物料的摩尔比为sio2/al2o3=15, na2o/sio2=1.07,h2o/sio2=21,再于35℃静置16小时得到导向剂。
[0061]
(3制备x分子筛
[0062]
在搅拌条件下,将69.44kg去离子水、3.74kg氢氧化钠、20.20kg(1)步制备的铝源、20.50kg的水玻璃、0.27kg(2)步制备的导向剂和0.16kg草酸钠加入反应釜中,得到x分子筛合成体系,其中各物料的摩尔比为 sio2/al2o3=2.95,na2o/sio2=1.76,h2o/sio2=80,草酸钠/al2o3=0.05,导向剂中所含al2o3与x分子筛合成体系中所含al2o3的摩尔比为0.15%。
[0063]
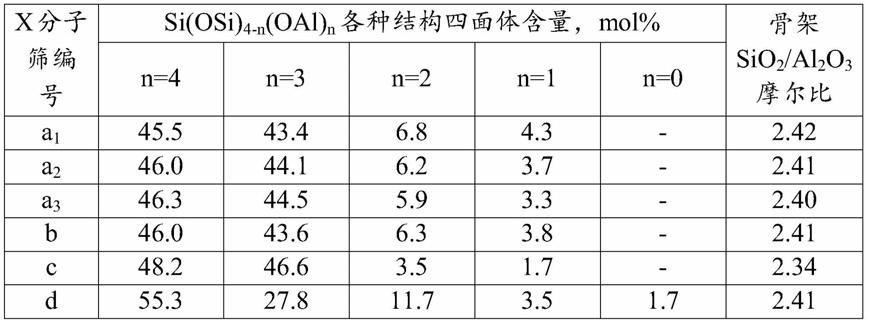
将上述分子筛合成体系转移到密闭反应釜中,于100℃水热晶化12小时,过滤,所得固体用去离子水洗涤至滤液ph=8~9,80℃干燥12小时,得到x分子筛a1,晶粒粒径为1.0微米,其xrd谱图见图1,
29
si固体核磁共振谱图见图2,从图2获得x分子筛a1骨架结构中各种结构四面体的含量及分子筛的骨架sio2/al2o3摩尔比,结果见表1。
[0064]
实例2
[0065]
按实例1的方法制备x分子筛,不同的是(3)步中,在搅拌条件下,将 69.44kg去离子水、3.74kg氢氧化钠、20.20kg铝源、20.50kg的水玻璃、0.27kg (2)步制备的导向剂和0.47kg草酸钠加入反应釜中,得到x分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=2.95,na2o/sio2=1.76,h2o/sio2=80,草酸钠/al2o3=0.15,导向剂中所含al2o3与x分子筛合成体系中所含al2o3的摩尔比为0.15%。将分子筛合成体系转移到密闭反应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得x分子筛a2,晶粒粒径为0.8微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架sio2/al2o3摩尔比见表1。
[0066]
实例3
[0067]
按实例1的方法制备x分子筛,不同的是(3)步中,在搅拌条件下,将 69.44kg去离子水、3.74kg氢氧化钠、20.20kg铝源、20.50kg的水玻璃、0.27kg (2)步制备的导向剂和0.79kg草酸钠加入到反应釜中,得到x分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=2.95,na2o/sio2=1.76,h2o/sio2=80,草酸钠/al2o3=0.25,导向剂中所含al2o3与x分子筛合成体系中所含al2o3的摩尔比为0.15%。导向剂中所含al2o3与x分子筛合成体系中所含al2o3的摩尔比为0.15%。将分子筛合成体系转移到密闭反应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得x分子筛a3,晶粒粒径为0.7微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架 sio2/al2o3摩尔比见表1。
[0068]
实例4
[0069]
按实例1的方法制备x分子筛,不同的是(3)步用0.68kg的酒石酸钠代替草酸钠,所得x分子筛合成体系中各物料的摩尔比为sio2/al2o3=2.95, na2o/sio2=1.76,h2o/sio2=80,酒石酸钠/al2o3=0.15,经水热晶化,过滤,用去离子水洗涤,干燥后得到x分子筛b,晶粒粒径为0.9微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架 sio2/al2o3摩尔比见表1。
[0070]
实例5
[0071]
按实例1的方法制备x分子筛,不同的是(3)步中,在搅拌条件下,将 69.36kg去离子水、4.07kg氢氧化钠、20.20kg铝源、20.50kg的水玻璃、0.27kg (2)步制备的导向剂和0.16kg草酸钠加入反应釜中,得到x分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=2.95,na2o/sio2=1.82,h2o/sio2=80,酒石酸钠/al2o3=0.05,导向剂中所含al2o3与x分子筛合成体系中所含al2o3的摩尔比为0.15%。将分子筛合成体系转移到密闭反应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得x分子筛c,晶粒粒径为0.6微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架 sio2/al2o3摩尔比见表1。
[0072]
对比例1
[0073]
按实例1的方法制备x分子筛,不同的是(3)步制备分子筛合成体系时,不加入草酸钠,得到x分子筛d,晶粒粒径为1.2微米,
29
si固体核磁共振谱见图3,从图3获得的其骨架结构中各种结构四面体的含量及分子筛的骨架 sio2/al2o3摩尔比见表1。
[0074]
实例6
[0075]
制备吸附剂并测试吸附性能。
[0076]
(1)滚球成型:取92千克(干基质量,下同)实例1制备的粉末状x分子筛a1与8千克高岭土(含90质量%的高岭石)混合均匀,放入转盘中边滚动边喷入适量的去离子水,使固体粉末聚集成小球,滚球时喷入的水量为固体粉末的8质量%。经筛分,取粒径为300~850μm的小球,80℃干燥10小时、540℃焙烧4小时。
[0077]
(2)原位晶化:将64千克(1)步焙烧后的小球置于200升氢氧化钠与氢氧化钾混合溶液中,混合溶液中氢氧根离子浓度为0.3mol/l,k/(na+k)摩尔比为0.2,于95℃原位晶化处理4小时,取晶化后固体水洗至洗涤液ph小于 10,80℃干燥10小时。
[0078]
(3)离子交换:将130毫升(2)步干燥后的小球装入离子交换柱中进行阳离子交换,用0.18m的硝酸钡和0.12m的氯化钾混合溶液以6.0小时-1
的体积空速于0.1mpa、94℃条件下连续交换8小时,硝酸钡溶液总用量为5000毫升,交换比为1.6。离子交换完成后,将固体于70℃用700毫升去离子水洗涤, 70℃氮气气氛干燥30小时,氮气气氛中于180℃脱水活化6小时,制得吸附剂 a,其组成和不同压力下的破碎率见表2。
[0079]
取1.0克吸附剂a,进行甲苯气相吸附实验测定其吸附容量以及灼基堆密度测试,结果见表2。
[0080]
取50毫升吸附剂a,进行液相脉冲实验测定其吸附选择性和px的吸附、解吸速率。
[0081]
解吸剂为对二乙苯时,实验所用解吸剂为30体积%的对二乙苯(pdeb) 和70体积%的正庚烷,脉冲进料液组成为各占5体积%的乙苯(eb)、对二甲苯(px)、间二甲苯(mx)、邻二甲苯(ox)、正壬烷(nc9)和75体积%的所述解吸剂。
[0082]
解吸剂为甲苯时,实验所用解吸剂为30体积%的甲苯(t)和70体积%的正庚烷,脉冲进料液组成为各占5体积%的乙苯(eb)、对二甲苯(px)、间二甲苯(mx)、邻二甲苯(ox)、正壬烷(nc9)和75体积%的所述解吸剂。其中正壬烷为示踪剂,正庚烷为稀释剂。
[0083]
分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0084]
实例7
[0085]
按实例6的方法制备吸附剂,不同的是(1)步中将实例2制备的x分子筛a2与高岭土混合后滚球成型,经原位晶化和离子交换得到吸附剂b,其组成、吸附容量、灼基堆密度、不
同压力下的破碎率、分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0086]
实例8
[0087]
按实例6的方法制备吸附剂,不同的是(1)步中将实例3制备的x分子筛a3与高岭土混合后滚球成型,经原位晶化和离子交换得到吸附剂c,其组成、吸附容量、灼基堆密度、不同压力下的破碎率、分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0088]
实例9
[0089]
按实例6的方法制备吸附剂,不同的是(1)步中将实例4制备的x分子筛b与高岭土混合后滚球成型,经原位晶化和离子交换得到吸附剂d,其组成、吸附容量、灼基堆密度、不同压力下的破碎率、分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0090]
实例10
[0091]
按实例6的方法制备吸附剂,不同的是(1)步中将实例5制备的x分子筛c与高岭土混合后滚球成型,经原位晶化和离子交换得到吸附剂e,其组成、吸附容量、灼基堆密度、不同压力下的破碎率、分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0092]
对比例2
[0093]
按实例6的方法制备吸附剂,不同的是(1)步中将对比例1制备的x分子筛d与高岭土混合后滚球成型,经原位晶化和离子交换得到吸附剂f,其组成、吸附容量、灼基堆密度、不同压力下的破碎率、分别以对二乙苯和甲苯为解吸剂测得的吸附性能见表2。
[0094]
实例11
[0095]
在连续逆流的小型模拟移动床上用吸附剂a进行分离对二甲苯实验,用对二乙苯为解吸剂。
[0096]
所用小型模拟移动床装置包括24根串联的吸附柱,每根柱长195毫米,柱内直径30毫米,吸附剂的总装填量为3300毫升。在串联的24根柱子首尾两端用循环泵连接构成一个封闭的环路,如图4所示。吸附原料、解吸剂、提取液、提余液四股进、出物料将24根吸附柱分成四个区段,即吸附原料(柱15) 和提余液(柱21)之间的7根吸附柱为吸附区,提取液(柱6)和吸附原料(柱 14)之间的9根吸附柱为提纯区,解吸剂(柱1)和提取液(柱5)之间的5 根吸附柱为解吸区,提余液(柱22)和解吸剂(柱24)之间的3根吸附柱为缓冲区。吸附分离操作的温度为177℃,压力为0.8mpa。
[0097]
操作过程中,分别以1680毫升/时和1750毫升/时的流量向上述模拟移动床中连续注入解吸剂对二乙苯和原料,并分别以630毫升/时和2800毫升/时的流量将提取液和提余液抽出装置。所述原料组成为:乙苯9.3质量%、对二甲苯18.5质量%、间二甲苯45.5质量%、邻二甲苯17.4质量%、非芳烃组分9.3 质量%。
[0098]
设定循环泵流量为4990毫升/时,每隔80秒四股物料同时向与液体流向相同的方向移动1根吸附柱(图4中,从实线至虚线位置,以此类推)。在稳定的操作状态下吸附剂a获得的对二甲苯的纯度为99.81质量%,收率为98.78 质量%。
[0099]
实例12
[0100]
在小型模拟移动床装置上装填吸附剂b,按实例11的方法进行吸附分离对二甲苯实验,稳定操作状态下获得的对二甲苯的纯度为99.84质量%,收率为 98.82质量%。
[0101]
对比例3
[0102]
在小型模拟移动床装置上装填对比吸附剂f,按实例11的方法进行吸附分离对二
甲苯实验,稳定操作状态下得到的对二甲苯的纯度为98.62质量%,收率为97.41质量%。
[0103]
实例13
[0104]
在连续逆流的小型模拟移动床上用吸附剂a进行分离对二甲苯实验,用甲苯为解吸剂。
[0105]
按实例11的方法从碳八芳烃中吸附分离对二甲苯,不同的是以甲苯为解吸剂,吸附分离操作的温度为135℃,压力为0.8mpa。
[0106]
操作过程中,分别以2118毫升/时和1925毫升/时的流量向模拟移动床中连续注入解吸剂甲苯和原料,并分别以1540毫升/时和2503毫升/时的流量将提取液和提余液抽出装置。
[0107]
设定循环泵流量为4890毫升/时,每隔80秒四股物料同时向与液体流向相同的方向移动1根吸附柱。在稳定的操作状态下吸附剂a获得的对二甲苯的纯度为99.76质量%,收率为97.85质量%。
[0108]
实例14
[0109]
在小型模拟移动床装置上装填吸附剂b,按实例13的方法进行吸附分离对二甲苯实验,稳定操作状态下获得的对二甲苯的纯度为99.78质量%,收率为 97.88质量%。
[0110]
对比例4
[0111]
在小型模拟移动床装置上装填对比吸附剂f,按实例13的方法进行吸附分离对二甲苯实验,稳定操作状态下得到的对二甲苯的纯度为96.55质量%,收率为92.50质量%。
[0112]
表1
[0113][0114]
n=4表示si(oal)4,n=3表示si(osi)(oal)3,n=2表示si(osi)2(oal)2,n=1 表示si(osi)3(oal),n=0表示si(osi)4[0115]
表2
[0116][0117]
*pdeb体系选择性—对二乙苯为解吸剂测得的吸附选择性,甲苯体系选择性—甲苯为解吸剂测得的吸附选择性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1