一种聚结型间二甲苯吸附剂及其制备方法与流程
1.本发明为聚结型分子筛吸附剂及其制备方法,具体地说,是一种聚结型间二甲苯吸附剂及其制备方法。
背景技术:
2.间二甲苯(mx)是重要的基本有机化工原料,广泛应用于合成树脂、农药、医药、涂料和染料等领域。工业上,高纯度的间二甲苯通常是采用吸附分离技术从含有乙苯、对二甲苯、间二甲苯和邻二甲苯的混合碳八芳烃中分离获得。
3.吸附剂是吸附分离技术的基础和核心,其活性组元多为沸石材料。cn1136549a和us6137024分别报道了以silicalite-1和氢型β沸石为活性组元的吸附剂,但是silicalite-1、β沸石的吸附容量较低,使其应用受到限制。相比而言,y分子筛的吸附容量较高,具有更为广阔的应用前景。
4.us4306107公开了一种从混合碳八芳烃中分离间二甲苯和乙苯的方法。该方法采用nay沸石为吸附剂的活性组元,以甲苯为解吸剂,利用nay沸石对间二甲苯的吸附能力最强、对二甲苯和邻二甲苯居中、乙苯最弱的特点,将混合碳八芳烃通入模拟移动床进行逆流操作,在模拟移动床的不同位置分别得到间二甲苯、对二甲苯和邻二甲苯、乙苯。
5.us4326092公开了一种从混合碳八芳烃中分离间二甲苯的方法,采用氧化硅与氧化铝摩尔比为4.5~5.0的nay沸石制备吸附剂,可以获得更高的间二甲苯选择性。
6.us5900523报道以氧化硅与氧化铝摩尔比为4.0~6.0的nay沸石为活性组元的吸附剂,水含量以500℃灼减量计为1.5~2.5质量%,以二氢化茚为解吸剂,在100~150℃进行液相吸附分离间二甲苯,取得了好的分离效果。
7.cn1939883a公开了一种从碳八芳烃异构体中分离间二甲苯的方法,采用氧化硅与氧化铝摩尔比为5~6的nay沸石制备吸附剂,该沸石含水0~8质量%,吸附温度25~250℃,解吸剂选自四氢化萘及其烷基化衍生物。
8.cn1050595c报道了一种以阳离子位同时被钠和锂离子占据的y沸石为活性组元的吸附剂,并用于从混合碳八芳烃中液相吸附分离间二甲苯,获得了更高的间二甲苯选择性。其中,锂离子占据沸石可交换位点的5%~35%,将吸附剂水含量以500℃灼减量计为1.5~3.0质量%。
技术实现要素:
9.本发明的目的是提供一种聚结型间二甲苯吸附剂及其制备方法,该吸附剂用于从混合碳八芳烃中吸附分离间二甲苯,具有较高的吸附选择性和吸附容量。
10.本发明提供的聚结型间二甲苯吸附剂包括96.0~99.0质量%的y分子筛和1.0~4.0质量%的基质,所述y分子筛由非转晶y分子筛和转晶生成的y分子筛组成,所述非转晶y分子筛的sio2/al2o3摩尔比为3.5~5.5,其骨架结构中si(oal)4与si(osi)1(oal)3结构四面体之和不大于12mol%,si(osi)2(oal)2和si(osi)3(oal)1结构四面体之和不小于81mol%,
si(osi)4结构四面体含量不大于7mol%。
11.本发明吸附剂活性组分y分子筛中的非转晶y分子筛的骨架结构中具有较多的si(osi)2(oal)2和si(osi)3(oal)1结构四面体,将其用于从混合碳八芳烃中吸附分离间二甲苯,可显著提高吸附剂的间二甲苯吸附选择性。
附图说明
12.图1为本发明实例1制备的y分子筛的xrd谱图。
13.图2为本发明实例1制备的y分子筛的
29
si固体核磁谱图。
14.图3为对比例1制备的y分子筛的
29
si固体核磁共振谱图。
15.图4为小型模拟移动床吸附分离示意图。
具体实施方式
16.y分子筛的骨架结构中si与si、si与al通过si-o-si、si-o-al相连,形成的四面体中含有五种不同的结构,可用通式si(osi)
4-n
(oal)
n
表示,通式中的n=0、1、2、3、4,其中含有
“-
o-al”键的四面体中,由于铝和硅的电性不同,导致分子筛具有吸附电性。通常,si-o-al的数量随着分子筛骨架硅/铝比的降低而升高。然而,在分子筛骨架硅/铝比基本相等的前提下,不同合成方法制备的y分子筛骨架结构中,si(osi)
4-n
(oal)
n
所示的各种四面体的含量存在差异。
17.本发明将具有特定骨架结构的y分子筛(非转晶y分子筛)与作为粘结剂的高岭土矿物、成型助剂和硅源混合后滚球成型,在高温下焙烧,使其中的高岭土转化为偏高岭土,然后再通过碱处理,使偏高岭土原位晶化转化成y分子筛。所述的具有特定骨架结构的y分子筛在合成过程中加入能和al形成配合物的多元羧酸根离子,其骨架中可形成较多的si(osi)2(oal)2和si(osi)3(oal)1结构四面体,si(osi)4结构四面体含量减少,制得的吸附剂具有较高的间二甲苯吸附选择性和吸附容量,可有效提高吸附分离装置的生产能力。
18.本发明所述吸附剂中含两种y分子筛,一种是非转晶y分子筛,具有特定的骨架结构,另一种是吸附剂成型过程中使用的粘结剂,一般为高岭土矿物与成型过程加入的硅源原位晶化后形成的y分子筛。所述吸附剂优选包括97.5~99.8质量%的y分子筛和0.2~2.5质量%的基质。
19.优选地,所述非转晶y分子筛骨架结构中,si(oal)4与si(osi)1(oal)3结构四面体之和为5~12mol%,si(osi)2(oal)2和si(osi)3(oal)1结构四面体之和为84~94mol%,si(osi)4结构四面体含量为0.1~5mol%。
20.所述非转晶y分子筛的晶粒粒径优选0.4~1.5微米,sio2/al2o3摩尔比优选为4~5。
21.吸附剂中所述的基质为高岭土矿物经原位晶化转晶后的剩余物。所述的高岭土矿物选自高岭石、地开石、珍珠石、耐火石和埃洛石中的至少一种。
22.本发明所述的吸附剂优选为小球状,小球的平均粒径优选300~850微米。
23.本发明所述吸附剂的制备方法,包括如下步骤:
24.(1)将非转晶nay分子筛、高岭土、硅源和成型助剂混合均匀,滚球成型制成小球,干燥后于530~600℃焙烧,所述非转晶nay分子筛与高岭土的质量比为85~94:6~15,
25.(2)将(1)步焙烧后所得小球用氢氧化钠或氢氧化钠与水玻璃的混合溶液于85~100℃进行原位晶化处理,使其中的高岭土原位晶化为y分子筛,然后水洗、干燥。
26.上述方法(1)步是将非转晶nay分子筛、高岭土、硅源与成型助剂混合后滚球成型,所述的高岭土中含有的晶化物质选自高岭石、地开石、珍珠石、耐火石、埃洛石或它们的混合物。所述高岭土中晶化物质的质量分数至少为90%。
27.所述的硅源选自正硅酸乙酯、硅溶胶、水玻璃、硅酸钠、硅胶和白炭黑中的一种或几种。优选地,加入的硅源中所含二氧化硅与高岭土的质量比为0.2~3.6、优选0.2~3.0。所述的成型助剂选自木质素、田菁粉、干淀粉、羧甲基纤维素和活性碳中的至少一种。成型助剂加量为固体粉料总量的1~6质量%。
28.(1)步所述的成型方法为滚球成型或喷雾成型。对于滚球成型方法,所用的设备可为转盘、糖衣锅或滚筒。滚球成型时,将混合均匀的固体粉料放入转动设备中,边滚动边喷水使固体粉料粘附团聚成小球。滚球时水用量为固体总质量的6~30%,优选6~20%。当加入的硅源为固体时,可与非转晶nay分子筛、高岭土混合;当加入的硅源为液体时,可与非转晶nay分子筛、高岭土混合,也可加入滚球成型所用的水中,或者既在固体粉料中加入硅源,又在水中加入硅源。
29.(1)步滚动成球后的小球,经过筛分,取一定范围粒径的小球,优选取粒径为300~850微米的小球,将其干燥、焙烧。所述干燥温度优选60~110℃,时间优选2~12小时,焙烧温度优选530~700℃,时间优选1.0~6.0小时。经过焙烧后,小球内的高岭土转化为偏高岭土,以便于(2)步转晶为nay分子筛。
30.上述方法(2)步为成型后小球的原位晶化,所述原位晶化可在氢氧化钠溶液或氢氧化钠与水玻璃的混合溶液中进行,原位晶化处理时的液/固比优选1.5~5.0升/千克,原位晶化处理温度优选85~100℃,时间优选0.5~8小时。
31.(2)步原位晶化处理所用的氢氧化钠溶液中氢氧根离子的浓度为0.1~3.0摩尔/升,优选0.5~1.5摩尔/升;当原位晶化处理使用氢氧化钠和水玻璃混合溶液时,其中氧化钠含量优选2~10质量%,二氧化硅含量优选1~6质量%。将原位晶化后的吸附剂水洗、干燥后即得球形吸附剂。所述的干燥温度优选70~110℃,干燥时间优选2~20小时。
32.本发明方法(1)步所述的非转晶nay分子筛的制备方法,包括如下步骤:
33.(
ⅰ
)将硅源、铝源、水和氢氧化钠混合,各物料摩尔比为sio2/al2o3=10~25,na2o/sio2=0.6~1.8,h2o/sio2=10~50,在0~60℃老化1~72小时,制成导向剂,
34.(
ⅱ
)将硅源、铝源、(1)步制备的导向剂、多元羧酸钠和水混合均匀形成分子筛合成体系,其中各物料的摩尔比为:sio2/al2o3=8.5~13.5,na2o/sio2=0.35~0.85,h2o/sio2=20~80,多元羧酸钠/al2o3=0.05~0.30,所加导向剂中所含al2o3与分子筛合成体系中所含al2o3的摩尔比为2~10%,
35.(
ⅲ
)将(
ⅱ
)步中的分子筛合成体系于90~150℃水热晶化8~48小时,晶化后所得固体经洗涤、干燥,得到y分子筛。
36.上述方法所述的铝源优选低碱度偏铝酸钠溶液、氧化铝、氢氧化铝、硫酸铝、氯化铝、硝酸铝和铝酸钠中的一种或几种,优选低碱度偏铝酸钠。所述低碱度偏铝酸钠溶液中al2o3含量为7~15质量%,na2o含量为7~20质量%。
37.所述的硅源选自正硅酸乙酯、硅溶胶、水玻璃、硅酸钠、硅胶和白炭黑中的一种或
几种,优选硅溶胶或水玻璃。
38.上述方法(
ⅰ
)步为合成导向剂,各物料摩尔比优选为sio2/al2o3=12~20,na2o/sio2=0.6~1.5,h2o/sio2=10~30,老化温度优选10~45℃,时间优选10~30小时。
39.上述方法(
ⅱ
)步制备分子筛合成体系,所述合成体系中,各物料的摩尔比优选为:sio2/al2o3=8.5~12,na2o/sio2=0.35~0.6,h2o/sio2=20~50,多元羧酸钠/al2o3=0.05~0.30,所加导向剂中所含al2o3与分子筛合成体系中所含al2o3的摩尔比优选为3~7%。所述的多元羧酸钠的碳原子数为2~5,含有的羧酸根为2~3个,或者上述多元羧酸钠的碳原子数为3~5,含有的羧酸根为2~3个,并且含有羟基。所述的不带羟基的多元羧酸钠可为草酸钠,带羟基的多元羧酸钠可为柠檬酸钠、酒石酸钠或苹果酸钠。所述的多元羧酸钠优选为草酸钠、柠檬酸钠、酒石酸钠和苹果酸钠中的一种或几种。
40.上述方法(
ⅲ
)步将分子筛合成体系进行水热晶化制备分子筛,所述的水热晶化温度优选90~130℃,时间优选10~40小时、更优选20~30小时。晶化后所得固体经洗涤后的干燥温度优选70~100℃,时间优选2~20小时。
41.本发明所述的吸附剂适用于从混合碳八芳烃中吸附分离间二甲苯。
42.吸附选择性和对吸附目的组分的吸附、解吸速率是评价吸附剂性能的重要指标。选择性为吸附平衡时,吸附相中两组分浓度的比值与非吸附相中该两组分浓度的比值之比。所述吸附平衡是指混合碳八芳烃与吸附剂接触后,吸附相和非吸附相之间不发生组分净转移时的状态。吸附选择性的计算公式如下:
[0043][0044]
其中,c和d表示欲进行分离的两种组分,a
c
和a
d
分别表示吸附平衡时吸附相中c、d两种组分的浓度,u
c
和u
d
分别表示吸附平衡时非吸附相中c、d两种组分的浓度。当两种组分的选择性β≈1.0时,表明吸附剂对两种组分的吸附能力相当,不存在被优先吸附的组分。当β大于或小于1.0时,表明一种组分被优先吸附。具体地说,当β>1.0时,吸附剂优先吸附c组分;当β<1.0时,吸附剂优先吸附d组分。从分离的难易程度讲,β值越大,吸附分离越容易进行。较快的吸附、解吸速率,有利于减少吸附剂和解吸剂的用量,提高产品收率,降低吸附分离装置的操作费用。
[0045]
本发明使用一种动态脉冲实验装置测定吸附选择性和间二甲苯的吸附、解吸速率。该装置由进料系统、吸附柱、加热炉、压力控制阀等组成。吸附柱为ф6
×
1800毫米的不锈钢管,吸附剂装量为50毫升。吸附柱下端入口与进料和氮气系统相连,上端出口接压力控制阀,再与流出物收集器连接。实验所用解吸剂组成为30体积%的甲苯(t)和70体积%的正庚烷(nc7),脉冲液组成为各占5体积%的乙苯(eb)、对二甲苯(px)、间二甲苯(mx)、邻二甲苯(ox)、正壬烷(nc9)和75体积%的上述解吸剂。
[0046]
吸附选择性的测定方法为:将称量好的吸附剂装入吸附柱震荡填实,在氮气流中于160~280℃脱水活化。然后通入解吸剂排除系统中的气体,将压力升至0.8mpa,温度升至145℃,停止通入解吸剂,按1.0小时
-1
的体积空速通入8毫升的脉冲进料液,然后停止通入脉冲液,并以同样的空速通入解吸剂进行脱附,每2分钟取3滴脱附液样品,用气相色谱分析组成。以脱附用解吸剂进料体积为横坐标,nc9和eb、px、mx、ox各组分浓度为纵坐标,绘制上述各组分的脱附曲线。作为示踪剂的nc9不被吸附,首先出峰,它给出了吸附系统的死体积。
将示踪剂半峰宽的中点作为零点,测定eb、px、mx、ox各组分半峰宽中点到零点的解吸剂进料体积,即净保留体积。两组分净保留体积之比即为吸附选择性β。如mx的净保留体积与eb的净保留体积之比即为mx相对于eb的吸附选择性,记为β
mx/eb
。
[0047]
为了实现吸附剂的循环连续使用,提取组分与解吸剂之间选择性也是一个重要的性能指标,可以通过对脉冲试验提取组分脱附曲线的进一步解析来确定。将mx的脉冲脱附曲线前沿流出液中mx浓度从10%上升到90%时所需的解吸剂体积定义为吸附速率[s
a
]
10-90
,将脱附曲线后沿mx浓度从90%下降到10%时所需的解吸剂体积定义为解吸速率[s
d
]
90-10
。二者的比值[s
d
]
90-10
/[s
a
]
10-90
即可表征为mx与解吸剂(t)之间的吸附选择性β
mx/t
。若β
mx/t
小于1.0,表示吸附剂对解吸剂的吸附能力太强,这对吸附过程是不利的,若β
mx/t
远大于1.0,则表示对解吸剂的吸附能力太弱,将会使脱附过程变得困难,理想的状况是β
mx/t
约等于或稍大于1.0。
[0048]
下面通过实例进一步说明本发明,但本发明并不限于此。
[0049]
实例和对比例中,吸附剂的物性参数测定方法如下:
[0050]
吸附剂抗压强度由一定压力下小球吸附剂的破碎率表示,破碎率越低,抗压强度越高。吸附剂抗压强度测定方法:采用dl-ii型颗粒强度测定仪(大连化工研究设计院生产)测定,吸附剂小球过300微米的筛后,在不锈钢筒体中装入约1.5毫升吸附剂。测定时安装一个与不锈钢筒体过盈配合的顶针,在预先设定好的压力下压一次后倒出吸附剂,再过300微米的筛称重,吸附剂加压测试前后质量减少量为在设定压力下吸附剂的破碎率。
[0051]
采用甲苯气相吸附实验测定吸附剂的吸附容量,具体操作方法为:在35℃下,使携带甲苯的氮气(甲苯分压为0.05mpa)与一定质量的吸附剂接触,直到甲苯达到吸附平衡。根据甲苯吸附前后吸附剂的质量差由下式计算出被测吸附剂的吸附容量。
[0052][0053]
其中,c为吸附容量,单位为毫克/克;m1为吸附甲苯前被测吸附剂的质量,单位为克;m2为吸附甲苯后被测吸附剂的质量,单位为克。
[0054]
吸附剂的灼基堆密度的测定方法:在100ml量筒中加入50ml吸附剂,在振实密度仪(辽宁仪表研究所有限责任公司生产)上振动5分钟,再加入50ml吸附剂并振动5分钟,量筒中吸附剂质量与体积之比为吸附剂堆密度;取一定质量的吸附剂于600℃灼烧2小时,并置于干燥器中冷却至室温,灼烧后与灼烧前吸附剂质量之比为灼基,灼基与吸附剂堆密度之积为灼基堆密度。
[0055]
实例1
[0056]
(1)制备铝源
[0057]
将200kg氢氧化铝、232.15kg氢氧化钠和652.33kg去离子水加入反应釜中,加热至100℃,搅拌6小时,形成澄清透明的低碱度偏铝酸钠溶液,作为铝源。所述铝源中al2o3含量为11.87质量%,na2o含量为16.59质量%,铝源的na2o与al2o3的摩尔比为2.3。
[0058]
(2)制备导向剂
[0059]
在搅拌条件下,将3.81kg氢氧化钠、8.86kg去离子水、4.48kg(1)步制备的铝源和23.24kg的水玻璃(水玻璃中sio2含量为20.17质量%,na2o含量为6.32质量%,下同)加入反应釜中,其各物料的摩尔比为sio2/al2o3=15,na2o/sio2=1.07,h2o/sio2=21,再于35℃静
置16小时得到导向剂。
[0060]
(3)制备y分子筛
[0061]
在搅拌条件下,将50.74kg的水玻璃、42.51kg去离子水、7.56kg(2)步制备的导向剂、8.66kg硫酸铝(含6.73质量%的al2o3)、11.01kg(1)步制备的低碱度偏铝酸钠溶液和0.13kg草酸钠加入反应釜中,其中各物料的摩尔比为sio2/al2o3=9.5,na2o/sio2=0.43,h2o/sio2=30,草酸钠/al2o3=0.05,导向剂中所含al2o3与y分子筛合成体系中所含al2o3的摩尔比为5%。
[0062]
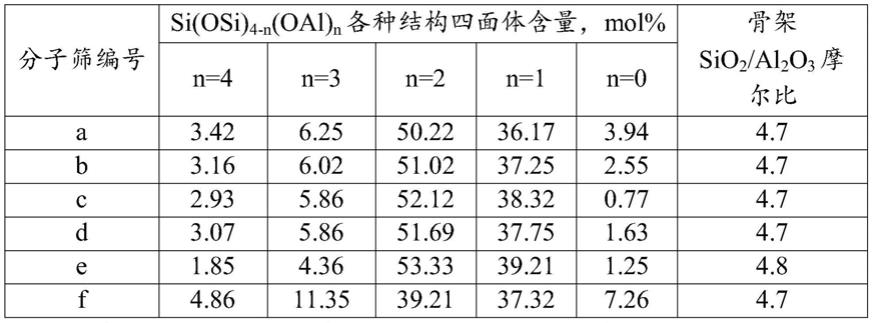
将上述分子筛合成体系转移到密闭反应釜中,100℃水热晶化28小时,过滤,所得固体用去离子水洗涤至滤液ph=8~9,80℃干燥12小时,得到y分子筛,编号为a,晶粒粒径为0.9微米,其xrd谱图见图1,
29
si固体核磁共振谱图见图2,从图2获得其骨架结构中各种结构四面体的含量及分子筛的骨架sio2/al2o3摩尔比见表1。
[0063]
实例2
[0064]
按实例1的方法制备y分子筛,不同的是(3)步中,在搅拌条件下,将50.74kg的水玻璃、42.51kg去离子水、7.56kg(2)步制备的导向剂、8.66kg硫酸铝、11.01kg(1)步制备的低碱度偏铝酸钠溶液和0.39kg草酸钠加入反应釜中,得到y分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=9.5,na2o/sio2=0.43,h2o/sio2=30,草酸钠/al2o3=0.15,导向剂中所含al2o3与y分子筛合成体系中所含al2o3的摩尔比为5%,将分子筛合成体系转移到密闭反应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得y分子筛b,晶粒粒径为0.8微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架sio2/al2o3摩尔比见表1。
[0065]
实例3
[0066]
按实例1的方法制备y分子筛,不同的是(3)步中,在搅拌条件下,将50.74kg的水玻璃、42.51kg去离子水、7.56kg(2)步制备的导向剂、8.66kg硫酸铝、11.01kg(1)步制备的低碱度偏铝酸钠溶液和0.65kg草酸钠加到反应釜中,得到y分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=9.5,na2o/sio2=0.43,h2o/sio2=30,草酸钠/al2o3=0.25,导向剂中所含al2o3与y分子筛合成体系中所含al2o3的摩尔比为5%,将分子筛合成体系转移到密闭反应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得y分子筛c,晶粒粒径为0.7微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架sio2/al2o3摩尔比见表1。
[0067]
实例4
[0068]
按实例1的方法制备y分子筛,不同的是(3)步用0.56kg的酒石酸钠代替草酸钠,得到y分子筛d,晶粒粒径为1.0微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架sio2/al2o3摩尔比见表1。
[0069]
实例5
[0070]
按实例1的方法制备y分子筛,不同的是(3)步中,在搅拌条件下,将50.74kg的水玻璃、40.96kg去离子水、7.56kg(2)步制备的导向剂、12.88kg硫酸铝、8.61kg(1)步制备的低碱度偏铝酸钠溶液和0.65kg草酸钠加到反应釜中,得到y分子筛合成体系,其中各物料的摩尔比为sio2/al2o3=9.5,na2o/sio2=0.35,h2o/sio2=30,草酸钠/al2o3=0.25,导向剂中所含al2o3与y分子筛合成体系中所含al2o3的摩尔比为5%,将分子筛合成体系转移到密闭反
应釜中,经水热晶化,过滤,用去离子水洗涤,干燥后得y分子筛e,晶粒粒径为1.2微米,采用
29
si固体核磁共振分析获得的骨架结构中各种结构四面体的含量及分子筛骨架sio2/al2o3摩尔比见表1。
[0071]
对比例1
[0072]
按实例1的方法制备y分子筛,不同的是(3)步制备分子筛合成体系时,不加入草酸钠,得到y分子筛f,晶粒粒径为1.0微米,
29
si固体核磁共振谱见图3,从图3获得的其骨架结构中各种结构四面体的含量及分子筛的骨架sio2/al2o3摩尔比见表1。
[0073]
表1
[0074][0075]
n=4表示si(oal)4,n=3表示si(osi)(oal)3,n=2表示si(osi)2(oal)2,n=1表示si(osi)3(oal),n=0表示si(osi)4[0076]
实例6
[0077]
制备本发明吸附剂并测试吸附性能。
[0078]
(1)滚球成型:将92千克(灼基质量,下同)实例1制备的nay分子筛a与8千克高岭土(含90质量%的高岭石)、3千克白炭黑、3千克田菁粉混合均匀,放入转盘中边滚动边喷入适量的去离子水,使固体粉料聚集成小球,滚球时喷入的水量为固体粉料的8质量%,白炭黑所含二氧化硅与高岭土的质量比为0.3。经筛分,取粒径为300~850μm的小球,80℃干燥10小时、540℃焙烧4小时。
[0079]
(2)原位晶化:将64千克(1)步焙烧后的小球置于200升氢氧化钠与水玻璃混合溶液中对焙烧后小球进行原位晶化处理,混合溶液中氧化钠含量为5质量%,二氧化硅含量为3质量%,于95℃原位晶化处理4小时,取晶化后固体水洗至洗涤液ph小于10,80℃干燥10小时,制得吸附剂a,吸附剂a中含98.5质量%的nay分子筛,1.5质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0080]
实例7
[0081]
按实例6的方法制备吸附剂,不同的是(1)步使用实例2制备的nay分子筛b与高岭土、白炭黑和田菁粉混合后滚球成型,经原位晶化制得吸附剂b,其中含98.2质量%的nay分子筛,1.8质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0082]
实例8
[0083]
按实例6的方法制备吸附剂,不同的是(1)步使用实例3制备的nay分子筛c与高岭土、白炭黑和田菁粉混合后滚球成型,经原位晶化制得吸附剂c,其中含99.0质量%的nay分
子筛,1.0质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0084]
实例9
[0085]
按实例6的方法制备吸附剂,不同的是(1)步使用实例4制备的nay分子筛d与高岭土、白炭黑和田菁粉混合后滚球成型,经原位晶化制得吸附剂d,其中含98.5质量%的nay分子筛,1.5质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0086]
实例10
[0087]
按实例6的方法制备吸附剂,不同的是(1)步使用实例5制备的nay分子筛e与高岭土、白炭黑和田菁粉混合后滚球成型,经原位晶化制得吸附剂e,其中含98.6质量%的nay分子筛,1.4质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0088]
对比例2
[0089]
按实例6的方法制备吸附剂,不同的是(1)步使用对比例1制备的nay分子筛f与高岭土、白炭黑和田菁粉混合后滚球成型,经原位晶化制得吸附剂f,其中含97.6质量%的nay分子筛,2.4质量%的基质,其吸附选择性、甲苯吸附容量、不同压力下的破碎率和灼基堆密度见表2。
[0090]
实例11
[0091]
在连续逆流的小型模拟移动床装置上用吸附剂a进行吸附分离间二甲苯的实验。
[0092]
所述小型模拟移动床装置包括24根串联的吸附柱,每根柱长195毫米,柱内直径30毫米,吸附剂的总装填量为3300毫升。在串联的24根柱子首尾两端用循环泵连接构成一个封闭的环路,如图4所示。吸附原料、解吸剂、提取液、提余液四股进、出物料将24根吸附柱分成四个区段,即吸附原料(柱15)和提余液(柱21)之间的7根吸附柱为吸附区,提取液(柱6)和吸附原料(柱14)之间的9根吸附柱为提纯区,解吸剂(柱1)和提取液(柱5)之间的5根吸附柱为解吸区,提余液(柱22)和解吸剂(柱24)之间的3根吸附柱为缓冲区。吸附分离的温度控制为145℃,压力为0.8mpa。
[0093]
操作过程中,分别按1600毫升/时和450毫升/时的流量连续地向上述模拟移动床装置中注入解吸剂甲苯和吸附原料,并以736毫升/时的流量将提取液抽出装置,1314毫升/时的流量将提余液抽出装置。所述吸附原料的组成为乙苯14.99质量%、对二甲苯20.14质量%、间二甲苯42.25质量%、邻二甲苯21.75质量%、非芳烃组分0.87质量%。设定循环泵流量3960毫升/时,每隔70秒四股物料位置按与液体流向相同的方向前移1根吸附柱(图4中,从实线至虚线位置,以此类推)。在稳定的操作状态下得到的间二甲苯纯度为99.64质量%,收率为96.85质量%。
[0094]
实例12
[0095]
在小型模拟移动床装置上装填吸附剂b,按实例11的方法进行吸附分离间二甲苯实验,稳定操作状态下获得的间二甲苯的纯度为99.65质量%,收率为97.36质量%。
[0096]
对比例3
[0097]
在小型模拟移动床装置上装填对比吸附剂f,按实例11的方法进行吸附分离间二甲苯实验,稳定操作状态下得到的间二甲苯的纯度为99.52质量%,收率为94.83质量%。
[0098]
表2
[0099]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1