一种金属单原子材料的制备方法与应用与流程
本发明属于化学催化技术领域,特别涉及一种金属单原子材料的制备方法与应用。
背景技术:
从2011年中国科学院大连化学物理研究所张涛院士提出利用金属单原子进行催化的想法以来(qiao,b.,etal.(2011).natchem3(8):634-641.),金属单原子因为具有超高催化活性及极高原子利用率而受到催化界从业者的广泛关注,经过近些年的发展,金属单原子催化剂已经迅速成为催化界的新兴方向和研究热点。目前制备金属单原子催化剂的方法主要有浸渍法、原子层沉积法、光电化学金属颗粒还原法等。但是这些方法仅适用于一种或者几种特定金属,同时这些制备方法还有过程比较复杂、制备成功率较低、可重复性较差等一系列亟待解决的问题。所以到目前为止,无法进行金属单原子催化剂的简单制备仍然是阻碍催化剂行业乃至整个工业生产发展的重大问题。而在金属单原子的制备过程中,由于需要对材料进行高温热解,所以导致金属前驱体生成金属单原子后会进一步生成金属纳米颗粒,进而使得制备得到的催化剂性能降低。对此,为了进一步推动金属单原子催化剂更广泛的研究和应用,亟需找到一种简单的方法对材料的制备方法进行改进,并需要进一步地研究出更为简单的、普适的方法用于大量制备金属单原子。
卟啉类金属化合物是构成自然界生命体的重要物质,血红素中的主要成分是卟啉铁,而叶绿素则主要由卟啉镁组成。与卟啉类金属化合物结构相似的,酞菁类金属化合物在1982年首次被发现,随后被用于作为染料。1964年,酞菁钴被发现可用于氧还原(r.jasinski,1964,nature,201:1212-3.),随后具有金属-非金属配位结构的这一类化合物进入了催化研究者的视野范围内。但是事实上,无论是卟啉类化合物还是酞菁类化合物,都需要和碳材料复合并热解后才能表现出良好的催化活性,而一般的优选热解温度为800℃及以上,而在这种温度下,该种金属化合物的结构变得不稳定,非常容易分解并形成纳米颗粒,并不能维持金属-非金属配位这种活性结构,以至于采用简单的复合并热解方法并不能完全将该种材料的优良性能充分发挥出来。所以亟需一种简单有效的方式保护热解过程中材料的金属-非金属这种活性结构。
技术实现要素:
为了克服上述现有技术的缺点,本发明的目的在于提供一种金属单原子材料的制备方法与应用,其技术路线简单并有效、具有普适性,可应用于电催化氧气还原反应及二氧化碳还原等各类型反应。
为了实现上述目的,本发明所提供的技术方案为:
一种金属单原子材料的制备方法,使用保护剂、添加剂以及与金属原子配位的前驱体,对含碳化合物进行一系列表面修饰处理,随后在惰性气氛下进行碳化,并后处理去除保护剂,得到金属单原子材料。
作为优选,所述与金属原子配位的前驱体包括卟啉类金属化合物、酞菁类金属化合物。优选为卟啉类金属化合物。
作为优选,所述保护剂为能够形成高温下稳定结构的化合物,如硅酸四乙酯,该种保护剂在添加剂加入的情况下会进一步形成在高温下具有稳定结构的化合物。
作为优选,所述添加剂为无机酸和/或有机酸,如盐酸、硫酸、硝酸等无机酸,甲酸、乙酸等有机酸,添加剂的作用为使保护剂进一步转化成具有高温稳定性的化合物。
作为优选,所述含碳化合物为碳单质和/或含碳有机物,如活性炭、纳米碳球、碳纳米管等碳单质,以及三聚氰胺、尿素、蔗糖、葡萄糖等含碳有机物,优选为纳米碳球。
作为优选,所述与金属原子配位的前驱体和含碳化合物之间的质量比在0.5-5之间,所述保护剂和添加剂之间的体积比可设置为1:0.5-1:5,优选0.5-5之间,最好为1:1。
作为优选,所述表面修饰处理过程为:将与金属原子配位的前驱体、含碳化合物进行溶剂混合,然后搅拌,溶剂蒸发,再加入保护剂和添加剂研磨;所述后处理过程为:对碳化后材料依次进行酸刻蚀、水洗、过滤、干燥、研磨。
作为优选,所述溶剂为四氢呋喃、n,n-二甲基甲酰胺(dmf)和二甲基亚砜(dmso)中的一种或者多种,优选的为n,n-二甲基甲酰胺(dmf),所述酸刻蚀中采用的酸为盐酸、硫酸、硝酸、氢氟酸中的一种或者几种。
作为优选,碳化的过程中,温度为500℃-1100℃,进一步优选为800℃;
作为优选,碳化时间为1小时~12小时,进一步优选为2小时。
作为优选,碳化的过程中,升温速率为1~10℃/分钟,优选为2℃/分钟。
作为优选,所述惰性气氛为氮气、氩气中的一种或者两种,纯度99-99.99%,优选的为99.99%。
所述金属单原子材料包括铁单原子材料、钴单原子材料、镍单原子材料、铜单原子材料。
本发明制备所得金属单原子材料可应用于电催化氧气还原反应及二氧化碳还原反应。
实验证实,与现有技术相比,本发明具有如下特点和优点:
(1)采用诸如卟啉类金属化合物、酞菁类金属化合物等作为金属前驱体能够轻松控制金属单原子负载量,便于研究者进一步改进材料性能。
(2)与已经报道的直接使用卟啉类、酞菁类金属化合物负载在碳基底的方法相比,本发明采用保护剂对卟啉、酞菁类金属化合物与碳基底的复合物进行包覆,一方面可以减少材料在高温热解这一条件下氮元素的流失,并进一步的抑制金属-非金属这种活性结构的破坏。另一方面由于金属原子在高温热解过程中容易因剧烈的热运动而导致纳米颗粒的形成,从而不能形成金属单原子材料,而本发明中金属前驱体受保护剂的空间位阻作用,阻止了高温碳化过程金属原子的聚集,并由此提高金属单原子的原子利用率。
(3)目前已经报道的金属单原子催化剂的制备方法往往只适用于某一种金属单原子材料的制备,同时制备路线复杂、可重复性不理想,而本发明提供的制备方法制备路线简单且具有普适性,可以制备任意金属单原子催化剂,例如fe单原子材料、co单原子材料、ni单原子材料和cu单原子材料等。
(4)利用本发明的方法制备的金属单原子能够均匀负载在不同类型的碳基底上,可作为电催化氧气还原、电催化二氧化碳还原等各类型反应的优良催化剂,能够表现出优异的催化活性和较优秀的原子使用效率。
综上,本发明选择含有金属-非金属配位结构的金属化合物,通过将其与保护剂、任意碳基底进行简单混合后再进行热解的处理方式,有效防止了催化活性结构的破坏,高效制备了金属单原子材料,并表现出良好的性能。该种技术路线简单并有效、具有普适性,具有良好的应用前景。
附图说明
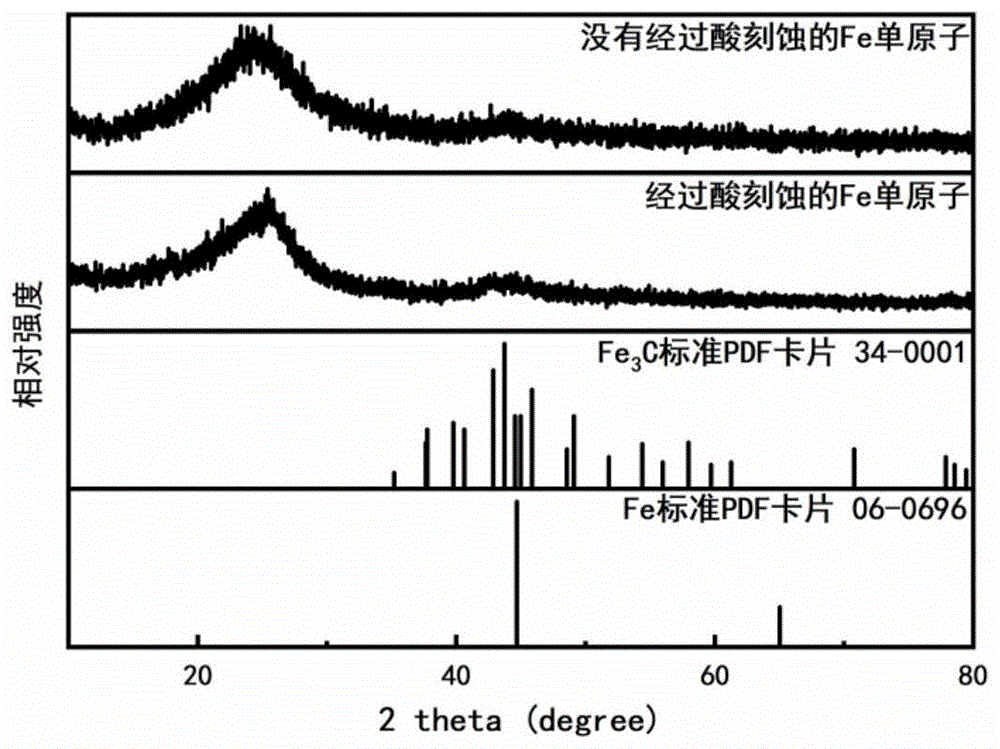
图1是本发明实施例1中制得的未经酸刻蚀的卟啉铁、导电炭黑复合粉末和经过酸刻蚀处理后的复合粉末的xrd对比。
图2是本发明实施例1中制得的fe单原子材料的sem的形貌图。
图3是本发明实施例1制得的fe单原子材料的stem-haadf的元素分布图。
图4是本发明实施例1制得的fe单原子材料的同步辐射exafs,其中fe箔和fe2o3作为参照对比。
图5是本发明实施例1中制得的fe单原子材料和传统商业pt/c的电化学氧气还原的线性扫描伏安法测试曲线。
图6是本发明实施例1中制得的fe单原子材料和传统商业pt/c的电化学氧气还原的抗甲醇测试曲线。
图7是本发明实施例2中制得的cu单原子材料的同步辐射exafs,其中cu箔和cuo作为参照对比。
具体实施方式
下面结合实施例与附图对本发明作进一步详细描述,需要指出的是,以下所述实施例旨在便于对本发明的理解,而对其不起任何限定作用。
实施例1:
制备fe单原子材料:称取质量比为1:2的卟啉铁、导电炭黑溶解于适当体积的n,n-二甲基甲酰胺(dmf)中,超声并搅拌24小时使之完全分散。随后放入鼓风干燥箱中进行溶剂蒸发。取完全干燥的样品置于玛瑙研钵当中,加入体积比为1:1的保护剂硅酸四乙酯和添加剂乙酸,随后研磨该种固液混合物直至完全干燥。将粉末放置到管式炉中,在氩气气氛中升温至800℃进行高温碳化2h,升温速率为2℃/min。然后自然降温至室温,取出得到黑色蓬松粉末。
将黑色粉末加入到适量体积的体积比为1:1的盐酸、氢氟酸的混合酸刻蚀液中进行搅拌刻蚀12h以除去保护剂,然后抽滤并用水洗涤5遍后将产物置于鼓风干燥箱中,样品完全干燥后得到fe单原子材料。
上述制得的fe单原子材料在酸刻蚀前后的xrd图谱如图1所示,可以发现在酸刻蚀前后材料的xrd衍射图谱基本没有变化,说明材料在加入保护剂的情况下经过高温热解后并没有fe3c杂质和fe纳米颗粒的生成。同时通过图2的sem电镜图可知,材料基本为20nm左右的纳米小颗粒。如图3所示,haddf-stem图片可以看到fe元素均匀分散于碳基底上。上述制得的fe单原子材料的同步辐射exafs如图4所示,其中fe箔和fe2o3作为参照对比,可以看出该fe单原子材料中没有fe-fe键,即不存在fe纳米颗粒。
上述制得的fe单原子材料的电化学氧气还原性能测试如下:
fe单原子材料液制备方法具体是:将6mg的fe单原子材料加入到200μl含有萘酚、水和乙醇混合溶液中(体积比为0.127:1:1),超声30分钟后得到均匀的黑色催化剂液。吸取10μl的该材料液滴到表面积为0.197cm2的旋转环盘电极上,在湿润的室温条件下干燥形成一层工作电极薄膜。测试时采用三电极电池测试,玻碳电极为工作电极,对电极为铂片电极,参比电极为ag/agcl电极,电解质为0.1mkoh,测试电压范围为0.15-1.05vvs.rhe。
为了比较,在相同测试条件下测试商业pt/c的电化学氧气还原性能。
测试结果如图5所示,商业pt/c的半波电位为0.86v,fe单原子材料的半波电位为0.89v。
同时如图6所示上述制得的fe单原子材料的抗甲醇能力突出
实施例2:
制备fe单原子材料:称取质量比为1:5的卟啉铁、葡萄糖溶解于适当体积的n,n-二甲基甲酰胺(dmf)中,超声并搅拌24小时使之完全分散。随后放入鼓风干燥箱中进行溶剂蒸发。取完全干燥的样品置于玛瑙研钵当中,加入体积比为1:1的保护剂硅酸四乙酯和添加剂盐酸,随后研磨该种固液混合物直至完全干燥。将粉末放置到管式炉中,在氩气气氛中升温至800℃进行高温碳化2h,升温速率为2℃/min。然后自然降温至室温,取出得到黑色蓬松粉末。
将黑色粉末加入到适量体积的体积比为1:1的硝酸、氢氟酸的混合酸刻蚀液中进行搅拌刻蚀12h以除去保护剂,然后抽滤并用水洗涤5遍后将产物置于鼓风干燥箱中,样品完全干燥后得到fe单原子材料。
实施例3:
制备fe单原子材料:称取质量比为1:3的酞菁铁、碳纳米管溶解于适当体积的n,n-二甲基甲酰胺(dmf)中,超声并搅拌24小时使之完全分散。随后放入鼓风干燥箱中进行溶剂蒸发。取完全干燥的样品置于玛瑙研钵当中,加入体积比为1:1的保护剂硅酸四乙酯和添加剂硝酸,随后研磨该种固液混合物直至完全干燥。将粉末放置到管式炉中,在氩气气氛中升温至800℃进行高温碳化2h,升温速率为2℃/min。然后自然降温至室温,取出得到黑色蓬松粉末。
将黑色粉末加入到适量体积的体积比为1:1的硫酸、氢氟酸的混合酸刻蚀液中进行搅拌刻蚀12h以除去保护剂,然后抽滤并用水洗涤5遍后将产物置于鼓风干燥箱中,样品完全干燥后得到fe单原子材料。
实施例4:
制备fe单原子材料:称取质量比为1:4的卟啉铁、尿素溶解于适当体积的n,n-二甲基甲酰胺(dmf)中,超声并搅拌24小时使之完全分散。随后放入鼓风干燥箱中进行溶剂蒸发。取完全干燥的样品置于玛瑙研钵当中,加入体积比为1:1的保护剂硅酸四乙酯和添加剂甲酸,随后研磨该种固液混合物直至完全干燥。将粉末放置到管式炉中,在氩气气氛中升温至800℃进行高温碳化2h,升温速率为2℃/min。然后自然降温至室温,取出得到黑色蓬松粉末。
将黑色粉末加入到适量体积的质量比分数为10%的氢氟酸中进行搅拌刻蚀12h以除去保护剂,然后抽滤并用水洗涤5遍后将产物置于鼓风干燥箱中,样品完全干燥后得到fe单原子材料。
实施例5:
制备cu单原子材料
本实施例中,cu单原子材料的制备方法如下与实施例1中的制备方法基本相同,所不同的是:用酞菁铜替代合成步骤中的卟啉铁,溶剂更换为二甲基亚砜(dmso),酸刻蚀液改为体积比为1:1的硝酸、氢氟酸的混合酸刻蚀液。
如图7所示,上述制得的cu单原子材料的同步辐射exafs结果与实施例1类似,其中cu箔和cuo作为参照对比,显示该cu单原子材料中没有cu-cu键,即不存在cu纳米颗粒,说明cu单原子的成功制备。
以上所述实施例对本发明的技术方案进行了详细说明,应理解的是以上所述仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!