一种广谱型血液灌流大孔吸附树脂及其制备方法与流程
本发明属于血液净化技术领域,具体涉及一种广谱型血液灌流大孔吸附树脂及其制备方法,适用于尿毒症、药物中毒、红斑狼疮、胆红素过高等疾病的治疗。
背景技术:
当代社会,随着人口老龄化趋势的不断增长,导致“三高”患者人数不断增加,由此引起的慢性肾病、肝病患者人数也在不断上升。流行病学研究发现肾衰竭、重症肝患者、药物中毒等患者人数正在持续增加,已经超出早期的预计。目前针对该类病种所采取的主要方式是血液透析,然而,尽管透析已有200多年历史且已成为血液净化领域的主要方式,但由透析引起的感染、发热、发痒、平衡失调等副作用仍不可忽视。血液净化领域中透析只能去除患者体内中小分子量的毒素,但对大分子量毒素几乎没有太大效果,且经常透析的患者会对血液透析产生强烈的依赖性,最终导致残余肾、肝功能更加低下。
不同于血液透析的是,血液灌流技术在去除患者体内小分子量毒素的同时,可完全有效的去除患者体内中大分子量毒素,更不会引起患者残余肾、肝功能持续低下。目前该技术已广泛应用于肝重症、肾病尿毒症、药物中毒等危重症领域。
血液灌流技术的原理:将血液借助体外循环动力装置引入装有固态吸附剂的灌流盒子中,通过吸附剂的吸附原理来清除血液中透析中不能清除的外源性或内源性毒素、药物或代谢废物。吸附剂是血液灌流装置的核心部件,其吸附性能直接关系治疗效果,而吸附剂的血液相容性及强度则直接关乎治疗的安全性。通常的吸附剂分为活性炭吸附剂、树脂炭化吸附剂、大孔树脂吸附剂,在这三种吸附剂中,最安全可靠的当属大孔树脂吸附剂。
大孔吸附树脂是通过苯乙烯-二乙烯苯悬浮聚合形成白球,白球经过氯甲基化形成氯球,再经傅克反应形成超高交联比表面积的大孔树脂。不同于活性炭、碳化树脂的是大孔树脂通过化学键合作用而形成,各分子间键合作用牢固,不易微粒脱落导致安全性能降低。活性炭及碳化树脂需经高温碳化,碳化后表面微粒易脱落,后期需要进行包膜处理。而大孔吸附树脂作为吸附剂,无需包膜即具备良好的机械强度及生物相容性。
目前市场存在的血灌流用大孔吸附树脂大多采用剧毒品氯甲醚直接对白球进行氯甲基化生成氯球,之后再将氯球用剧毒有机溶剂硝基苯溶胀后在路易斯酸催化下进行二次交联而制得。其制备过程中所用的溶胀剂硝基苯属于剧毒易爆溶剂,受公安部门管制,且硝基苯高热爆炸,对环境具有不可逆危害。
技术实现要素:
本发明的目的是提供一种溶剂残留及生物相容性等性能方面良好,且制备过程安全的广谱型血液灌流大孔吸附树脂的制备方法。
本发明的技术方案是提供了一种广谱型血液灌流大孔吸附树脂的制备方法,包括如下步骤:
1)白球聚合步骤,将苯乙烯类单体、多乙烯基类单体、致孔剂和引发剂混合均匀形成油相,再将该油相在分散介质中进行悬浮聚合生成聚合白球基体;
2)一锅法制备氯球步骤,将甲缩醛、多聚甲醛、氯化亚砜反应后再加入上述聚合白球基体及催化剂进行氯甲基化反应生成氯球;
3)氯球嫁接官能团步骤,在上述氯球中加入溶胀剂溶胀反应后再加入官能团试剂、催化剂进行傅克反应生成粗树脂,所述溶胀剂为二氯乙烷;
4)后处理步骤,将上述粗树脂用甲缩醛进行提取、水煮,之后加入乙醇搅拌升温至80℃,回流2小时后抽滤乙醇,再加入纯水升温100℃蒸馏去除树脂孔道残留乙醇,之后滤除水分,即得大孔吸附树脂。
进一步的,所述步骤1)中,悬浮聚合反应的定型反应温度为65~95℃,定型反应时间为3~15h,且控制悬浮聚合反应所生成的聚合白球基体粒度为0.4~0.8mm。
进一步的,所述步骤1)中,聚合白球基体经甲缩醛提取、水煮、烘干至含水量≤2%。
进一步的,所述苯乙烯类单体为苯乙烯,多乙烯基类单体为二乙烯苯,且二乙烯苯占两种单体总质量的3~15%;所述致孔剂为二甲苯、汽油、白油或液体石蜡,且致孔剂加入量为两种单体总质量的50~150%;所述引发剂为bpo或过氧化十二烷酰,且引发剂加入量为两种单体总质量的0.5~2%;所述分散介质为0.1%次甲基蓝、明胶配置液、5%羧甲基纤维素、自来水在40℃搅拌混合均匀形成的水相,且分散介质与油相的体积比为1:1~5:1。
进一步的,所述步骤2)中,待多聚甲醛、甲缩醛同滴加的氯化亚砜反应完全的料液氯含量≥40%时,再加入聚合白球基体及催化剂,且生成的氯球中氯含量≥20%;所述催化剂为氯化锌、氯化铁或氯化铝。
进一步的,所述步骤3)中,所述官能团试剂为含有羧基、羟基、硝基或磺酸基的极性基团的化合物,且官能团试剂的摩尔质量占氯球平均摩尔质量的1~10%;所述催化剂为氯化锌或氯化铝,且催化剂加入量占氯球质量的50~150%;所述溶胀剂加入量占氯球质量的100~800%。
进一步的,所述官能团试剂为对磺酸基苯甲酸、对甲基苯磺酸、对硝基酚、水杨酸或磺基水杨酸。
具体的,所述步骤1)中,调整搅拌速度,待油相在分散介质中形成均匀粒度液滴且粒径范围0.4~0.8mm时,升温至60~65℃进行固化定型,反应时间3小时到6小时;再升温75~80℃,固化定型1小时到3小时;最后升温90~95℃继续固化定型6小时到9小时后停止反应;然后用6倍聚合白球基体体积的甲缩醛进行提取,之后于100℃下进行煮球去除聚合白球基体孔道残留的甲缩醛,再抽滤去除聚合白球基体表层水分于105℃下烘干两小时,测得聚合白球基体水分含量≤2%即可;
所述步骤2)中,待多聚甲醛、甲缩醛同滴加的氯化亚砜反应完全,料液氯含量≥40%时,将步骤1)中烘干的聚合白球基体投入该反应后的料液中溶胀四小时,加入氯化锌或氯化铁作为催化剂,升温42℃回流反应10小时到15小时至氯球中氯含量≥20%为止;抽滤母液去回收,氯球用3倍氯球体积的甲醇进行洗涤后于60℃下烘干;
所述步骤3)中,在上述步骤2)所得氯球中加入氯球4~8倍体积的二氯乙烷作为溶胀剂于室温下溶胀4小时,之后加入官能团试剂和催化剂后搅拌1小时,缓慢升温80℃反应6小时,降室温抽出母液后,加入纯水于100℃下蒸馏来去除树脂孔道残留的二氯乙烷,再抽滤收集该树脂去进行后处理;
所述步骤4)中,将步骤3)制备的树脂用3倍体积的甲缩醛进行提取、水煮,之后加入3倍树脂体积的医用酒精搅拌升温至80℃,回流2小时后抽滤乙醇,再加入4倍树脂体积的纯水,升温100℃蒸馏去除树脂孔道残留乙醇,之后滤除水分再检测大孔吸附树脂的各项指标。
另外,本发明还提供了一种采用上述制备方法制得的大孔吸附树脂。
进一步的,所述大孔吸附树脂的比表面积为900~1350m2/g,孔体积范围为1.5~2.0cm3/g,平均孔径范围15~55nm,粒径范围0.4~1.2mm。
与现有技术相比,本发明的有益效果:
(1)本发明提供的这种广谱型血液灌流大孔吸附树脂的制备方法在白球氯甲基化阶段采用一锅法制备氯球,直接将甲缩醛、多聚甲醛、氯化亚砜反应一定时间后加入白球、催化剂,再进行氯甲基化制备氯球,避免了传统工艺中直接使用剧毒品氯甲醚的危害,且用该法所制备的干氯球进行傅克反应二次交联时,所生成的树脂产品中基本无残氯存在,同时交联过程使用二氯乙烷作为溶胀剂,无需使用易爆高毒品硝基苯作为溶胀剂,生产安全。
(2)本发明提供的这种广谱型血液灌流大孔吸附树脂的制备方法相比于活性炭或树脂基活性炭,无需包膜即可具有可靠的强度和良好的生物相容性,且在高比表面积、合适的孔融孔径等满足物理吸附的条件下,加入官能团试剂所带入的极性基团也可和血液毒素靶分子间形成化学盐桥,促进树脂与靶细胞分子间的特异性吸附。
(3)本发明提供的这种广谱型血液灌流大孔吸附树脂的制备方法制备的大孔吸附树脂经国家标准《yy/t0464-2019》所要求方法检测,结果表明其对肌酐的吸附率达到90%以上,对vb12吸附率达到95%以上,对戊巴比妥吸附率95%以上;而且用颗粒强度检测仪对其强度进行检测,测得破碎强度为8n左右,磨后圆球率达到85%以上,溶剂残留经gb/t24396-2009检测≤2ppm,且生物相容性等性能方面相较于市场上存在的血液灌流用吸附材料均有所改善。
以下将结合附图对本发明做进一步详细说明。
附图说明
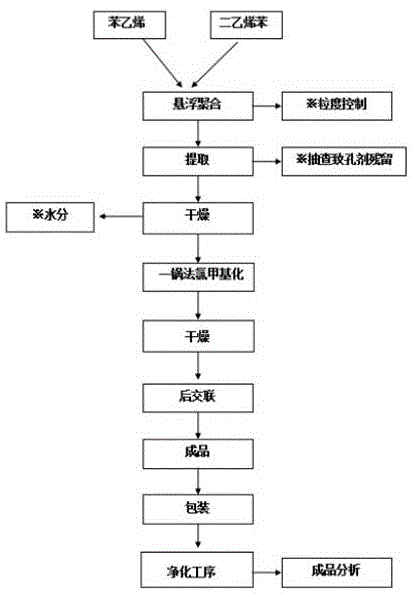
图1是本发明广谱型血液灌流大孔吸附树脂的制备方法的工艺流程图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
如图1所示,在本发明的实施例中提供了一种广谱型血液灌流大孔吸附树脂的制备方法,具体过程如下:
(1)白球聚合步骤,将苯乙烯类单体、多乙烯基类单体、致孔剂和引发剂混合均匀形成油相,再将该油相在分散介质中进行悬浮聚合生成聚合白球基体。其中,所述苯乙烯类单体为苯乙烯,多乙烯基类单体为二乙烯苯,且二乙烯苯占两种单体总质量的3~15%;所述致孔剂为二甲苯、汽油、白油或液体石蜡,且致孔剂加入量为两种单体总质量的50~150%;所述引发剂为bpo或过氧化十二烷酰,且引发剂加入量为两种单体总质量的0.5~2%;所述分散介质为0.1%次甲基蓝、明胶配置液、5%羧甲基纤维素、自来水在40℃搅拌混合均匀形成的水相,且分散介质与油相的体积比为1:1~5:1。
聚合反应原理如下:
在优选的实施例中,悬浮聚合反应的定型反应温度为65~95℃,定型反应时间为3~15h,且控制悬浮聚合反应所生成的聚合白球基体粒度为0.4~0.8mm。具体操作时可按下面步骤进行,调整搅拌速度,待油相在水相中形成均匀粒度液滴且粒径范围0.4~0.8mm时,升温至60~65℃进行固化定型,反应时间3小时到6小时;再升温75~80℃,固化定型1小时到3小时;最后升温90~95℃继续固化定型6小时到9小时后停止反应;然后用6倍聚合白球基体体积的甲缩醛进行提取,之后于100℃下进行煮球去除聚合白球基体孔道残留的甲缩醛,再抽滤去除聚合白球基体表层水分于105℃下烘干两小时,测得聚合白球基体水分含量≤2%即可。
(2)一锅法制备氯球步骤,将甲缩醛、多聚甲醛、氯化亚砜反应后再加入上述聚合白球基体及催化剂进行氯甲基化反应生成氯球。
氯球制备原理如下:
具体操作过程如下,待多聚甲醛、甲缩醛同滴加的氯化亚砜反应完全,料液氯含量≥40%时,将步骤(1)中烘干的聚合白球基体投入该反应后的料液中溶胀4小时,加入氯化锌或氯化铁作为催化剂,升温42℃回流反应10小时到15小时至氯球中氯含量≥20%为止;抽滤母液去回收,氯球用3倍氯球体积的甲醇进行洗涤后于60℃下烘干。
(3)氯球嫁接官能团步骤,在上述氯球中加入溶胀剂溶胀反应后再加入官能团试剂、催化剂进行傅克反应生成粗树脂。其中,所述官能团试剂为含有羧基、羟基、硝基或磺酸基的极性基团的化合物,如对磺酸基苯甲酸、对甲基苯磺酸、对硝基酚、水杨酸或磺基水杨酸,官能团试剂的摩尔质量占氯球平均摩尔质量的1~10%,优选3~5%;所述催化剂为氯化锌或氯化铝,且催化剂加入量占氯球质量的50~150%;所述溶胀剂为二氯乙烷,二氯乙烷作为溶胀剂可通过蒸馏的方式进行回收,比易爆品硝基苯作为溶胀剂回收时更加安全,溶胀剂加入量占氯球质量的100~800%,优选500%。
氯球同官能团试剂进行傅克反应时,自身氯甲基上的氯元素也会和自身的氢元素发生傅克反应,具体反应方程式如下
注:hr为官能团试剂。
具体操作过程如下,在上述步骤(2)所得氯球中加入氯球4~8倍体积的二氯乙烷作为溶胀剂于室温下溶胀4小时,之后加入官能团试剂和催化剂后搅拌1小时,缓慢升温80℃反应6小时,降室温抽出母液后,加入纯水于100℃下蒸馏来去除树脂孔道残留的二氯乙烷,再抽滤收集该树脂去进行后处理。
(4)后处理步骤,将上述粗树脂装入提取柱,用3倍等体积的甲缩醛进行提取,之后树脂导入另外干净反应釜中加纯水煮球去除孔道残余甲缩醛,再加入3倍体积的医用酒精搅拌升温至80℃,回流2小时后抽滤乙醇,再加入适量纯水升温100℃蒸馏去除树脂孔道残留乙醇,之后滤除水分,即得大孔吸附树脂,大孔吸附树脂按照《yy/t0464-2019》所要求方法检测各项指标。
经过对大孔吸附树脂相关参数的表征,表明本发明实施例制备的大孔吸附树脂的比表面积为900~1350m2/g,孔体积范围为1.5~2.0cm3/g,平均孔径范围15~55nm,粒径范围0.4~1.2mm。
下面通过具体实施例说明本发明广谱型血液灌流大孔吸附树脂的制备方法及制备的大孔吸附树脂的性能。
实施例1:
步骤(1)白球聚合步骤:
在1000ml反应釜中加0.1%次甲基蓝3.3ml、明胶5g、羧甲基纤维素1.25g、自来水400ml,开启搅拌温度升至40℃,直至反应釜内液混合均匀形成水相;在1000ml烧杯中加入苯乙烯181g,63.3%的二乙烯苯19g、二甲苯200g、bpo2g后于室温下搅拌均匀形成油相,再将油相由烧杯在室温下倒入水相反应釜,于室温下静置分层,再缓慢开启搅拌调整粒度0.4~0.8mm之间,30分钟升温80℃保温3小时,之后于30分钟升至90℃保温3小时,之后30分钟升温95℃保温6小时后降室温,得到聚苯乙烯-二乙烯苯基的白球,白球再经6倍体积的甲缩醛提取,用4倍体积纯水于100℃下煮球后烘干至含水量≤2%即可。
步骤(2)一锅法制备氯球步骤:
1000ml反应釜中加入多聚甲醛45g、甲缩醛90ml、开启搅拌于室温下缓慢滴加56ml工业盐酸,滴毕,再缓慢滴加286ml氯化亚砜,3小时滴毕(废气经喷淋吸收塔),之后升温42℃回流10小时,测料液氯含量≥40%即可;降至室温,将100g干白球投入反应釜中搅拌溶胀4小时,加入100g氯化锌后缓慢升温42℃,反应至所得到的氯球氯含量≥20%即可,不达标则继续反应。反应结束后,抽滤分离母液去回收,将所得氯球用4倍体积的甲醇洗涤3次后于60℃下烘干。
步骤(3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的二氯乙烷于室温下溶胀4小时,之后加入3g对磺酸基苯甲酸试剂、100g氯化铁后于1小时升温80℃反应6小时,降温抽出母液后加入4倍体积的纯水于100℃下蒸馏除去树脂孔道残留有机溶剂,之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:
将步骤(3)所得树脂装入提取柱用3倍树脂体积的甲缩醛进行过柱提取,之后将树脂导入另干净2000ml反应釜中加四倍树脂体积的纯水于100℃下煮球去除孔道残余甲缩醛。水煮后的大孔吸附树脂,加入3倍体积的乙醇搅拌缓慢升温至80℃,回流两小时后抽滤乙醇。再加入4倍树脂体积的纯水升温100℃蒸馏去除树脂孔道残留乙醇,再滤除水分,收集处理后的树脂去检测。
本实施例制得的广谱型血液灌流吸附树脂的外观为黄褐色颗粒状,按照《yy/t0464-2019》所要求方法检测本实施例广谱型血液灌流吸附树脂各项指标,结果表明其对肌酐的吸附率88%,对vb12吸附率96.1%,对戊巴比妥吸附率95.3%以上。该树脂色泽呈深棕色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为8.3n,根据《gb/t12598-2001》测得磨后圆球率85%。
实施例2:
步骤(1)白球聚合步骤:
在1000ml反应釜中加0.1%次甲基蓝3.3ml、明胶5g、羧甲基纤维素1.25g、自来水400ml,开启搅拌温度升至40℃,直至反应釜内液混合均匀形成水相;在1000ml烧杯中加入苯乙烯181g,63.3%的二乙烯苯19g、白油200g、bpo2g后于室温下搅拌均匀形成油相,再将油相由烧杯在室温下倒入水相反应釜,于室温下静置分层,再缓慢开启搅拌调整粒度0.4mm~0.8mm之间,30分钟升温80℃保温3小时,之后于30分钟升至90℃保温3小时,之后30分钟升温95℃保温6小时后降室温,得到聚苯乙烯-二乙烯苯基的白球,白球经6倍体积的甲缩醛提取后,再用4倍体积纯水于100℃下煮球后烘干至含水量≤2%即可。
步骤(2)一锅法制备氯球步骤:该步骤同实例(1)中步骤(2)。
步骤(3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的二氯乙烷于室温下溶胀4小时,之后加入3g水杨酸试剂、100g氯化铁后于1小时升温80℃反应6小时,降温抽出母液后加入4倍体积的纯水于100℃下蒸馏除去树脂孔道残留有机溶剂。之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:该步骤同实例(1)中步骤(4)。
本实施例制得的广谱型血液灌流吸附树脂的外观为红褐色颗粒状,按照《yy/t0464-2019》所要求方法检测本实施例广谱型血液灌流吸附树脂各项指标,结果表明其对肌酐的吸附率86.5%,对vb12吸附率96.1%,对戊巴比妥吸附率95.1%以上。该树脂色泽呈深褐色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为8.5n,根据《gb/t12598-2001》测得磨后圆球率83%。
实施例3:
步骤(1)白球聚合步骤:
在1000ml反应釜中加0.1%次甲基蓝3.3ml、明胶5g、羧甲基纤维素1.25g、自来水400ml,开启搅拌温度升至40℃,直至反应釜内液混合均匀形成水相;在1000ml烧杯中加入苯乙烯181g、63.3%二乙烯苯19g、汽油200g、bpo2g后于室温下搅拌均匀形成油相,再将油相由烧杯在室温下倒入水相反应釜,于室温下静置分层,再缓慢开启搅拌调整粒度0.4mm~0.8mm之间,30分钟升温80℃保温3小时,之后于30分钟升至90℃保温3小时,之后30分钟升温95℃保温6小时后降室温,得到聚苯乙烯-二乙烯苯基的白球,白球再经6倍体积的甲缩醛提取后,再用4倍体积纯水于100℃下煮球后烘干至含水量≤2%即可。
步骤(2)一锅法制备氯球步骤:该步骤同实例(1)中步骤(2)。
步骤(3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的二氯乙烷于室温下溶胀4小时,之后加入3g对甲基苯磺酸试剂、80g氯化铝后于1小时升温80℃反应6小时,降温抽出母液后加入4倍体积的纯水于100℃下蒸馏除去树脂孔道残留有机溶剂。之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:该步骤同实例(1)中步骤(4)。
本实施例制得的广谱型血液灌流吸附树脂的外观为黄褐色颗粒状,按照《yy/t0464-2019》所要求方法检测本实施例广谱型血液灌流吸附树脂各项指标,结果表明其对肌酐的吸附率88%,对vb12吸附率95.5%,对戊巴比妥吸附率95.5%以上。该树脂色泽呈深棕色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为9n,根据《gb/t12598-2001》测得磨后圆球率86%。
实施例4:
步骤(1)白球聚合步骤:
在1000ml反应釜中加0.1%次甲基蓝3.3ml、明胶5g、羧甲基纤维素1.25g、自来水400ml,开启搅拌温度升至40℃,直至反应釜内液混合均匀形成水相;在1000ml烧杯中加入苯乙烯181g、63.3%二乙烯苯19g、甲苯200g、bpo2g后于室温下搅拌均匀形成油相,再将油相由烧杯在室温下倒入水相反应釜,于室温下静置分层,再缓慢开启搅拌调整粒度0.4mm~0.8mm之间,30分钟升温80℃保温3小时,之后于30分钟升至90℃保温3小时,之后30分钟升温95℃保温6小时后降室温,得到聚苯乙烯-二乙烯苯基的白球,白球再经6倍体积的甲缩醛提取后,再用4倍体积纯水于100℃下煮球后烘干至含水量≤2%即可。
步骤(2)一锅法制备氯球步骤:该步骤同实施例(1)中步骤(2)。
步骤(3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的二氯乙烷于室温下溶胀4小时,之后加入3g磺基水杨酸试剂、100g氯化铁后于1小时升温80℃反应6小时,降温抽出母液后加入4倍体积的纯水于100℃下蒸馏除去树脂孔道残留有机溶剂,之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:该步骤同实例(1)中步骤(4)。
本实施例制得的广谱型血液灌流吸附树脂的外观为深红棕色颗粒状,按照《yy/t0464-2019》所要求方法检测本实施例广谱型血液灌流吸附树脂各项指标,结果表明其对肌酐的吸附率90%,对vb12吸附率95.4%,对戊巴比妥吸附率95%。该树脂色泽呈红棕色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为8.8n,根据《gb/t12598-2001》测得磨后圆球率87%。
实施例5:
步骤(1)白球聚合步骤:该步骤同实施例(1)中步骤(1)。
步骤(2)一锅法制备氯球步骤:该步骤同实施例(1)中步骤(2)。
步骤3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的二氯乙烷于室温下溶胀4小时,之后加入3g对硝基苯酚试剂、80g氯化铝后于1小时升温80℃反应6小时,降温抽出母液后加入4倍体积的纯水于100℃下蒸馏除去树脂孔道残留有机溶剂,之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:该步骤同实施例(1)步骤(4)。
本实施例制得的广谱型血液灌流吸附树脂的外观为绿褐色颗粒状,按照《yy/t0464-2019》所要求方法检测本实施例广谱型血液灌流吸附树脂各项指标,结果表明其对肌酐的吸附率87%,对vb12吸附率94.1%,对戊巴比妥吸附率95.3%以上。该树脂色泽呈绿褐色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为8.1n,根据《gb/t12598-2001》测得磨后圆球率88%。
对比例:
本对比例中广谱型血液灌流大孔吸附树脂的制备过程与上述实施例1大致相同,不同之处在于在步骤(1)中,将单体交联度改为11%,且以白油作为致孔剂来聚合反应成白球。在步骤(2)中采用氯甲醚直接法制备氯球,同时在步骤(3)中采用硝基苯作溶胀剂,具体过程如下:
步骤(1)白球聚合步骤:在1000ml反应釜中加0.1%次甲基蓝3.3ml、明胶5g、羧甲基纤维素1.25g、自来水400ml,开启搅拌温度升至40℃,直至反应釜内液混合均匀形成水相;在1000ml烧杯中加入苯乙烯165.24g,含量63.3%的二乙烯苯34.76g、致孔剂白油300g、引发剂bpo2g后于室温下搅拌均匀形成油相,再将油相由烧杯在室温下倒入水相反应釜,于室温下静置分层,再缓慢开启搅拌调整粒度0.4~0.8mm之间,30分钟升温80℃保温3小时,之后于30分钟升至90℃保温3小时,之后30分钟升温95℃保温6小时后降室温,得到聚苯乙烯-二乙烯苯基的白球,白球再经6倍体积的甲缩醛提取,用4倍体积纯水于100℃下煮球后烘干至含水量≤2%即可。
步骤(2)氯甲醚直接法制备氯球步骤:
1000ml反应釜中加入500ml氯甲醚、100g干白球投入反应釜中搅拌室温下溶胀4小时,加入100g氯化锌后缓慢升温42℃,反应至所得到的氯球氯含量≥18%即可,不达标则继续反应。反应结束后,抽滤分离母液去回收,将所得氯球用4倍体积的乙醇洗涤3次后于60℃下烘干。
步骤(3)氯球傅克反应嫁接官能团步骤:
1000ml反应釜中加入步骤(2)所得干氯球100g,加入5倍体积的硝基苯于室温下溶胀4小时,之后加入100g氯化锌后于1小时升温80℃反应6小时,降温抽滤出母液后加入4倍体积的乙醇升温60℃搅拌1小时,再抽滤除去树脂孔道残留有机溶剂,之后抽滤收集该材料进行后处理。
步骤(4)后处理步骤:该步骤同实施例(1)步骤(4)。
本实施例制得的广谱型血液灌流吸附树脂的为棕黑色颗粒状,按照《yy/t0464-2019》所要求方法检测本实血液灌流用吸附树脂各项指标,结果表明其对肌酐的吸附率83.6%,对vb12吸附率95.1%,对戊巴比妥吸附率93.5%以上。该树脂色泽呈棕黑色,表面光滑,用颗粒强度检测仪按照有关标准对其强度进行检测,测得破碎强度为7.3n,磨后圆球率根据《gb/t12598-2001》测得85%。
上述实施例1~5得到的产品经净化处理后按照gb/t24396-2009检测成品的溶剂残留均低于2ppm,而上述对比例得到的产品经净化处理后的溶剂残留量则高于10ppm。究其原因,主要由于对照例采用的沸点210.9℃的硝基苯作溶胀剂,不同于实施例采取的低沸点83.7℃的二氯乙烷,本发明所采用的二氯乙烷溶胀剂可在后期抽滤后加水蒸馏去除。而硝基苯溶胀剂由于沸点过高,且遇高热易爆等特点不易采取蒸馏去除的方式,最终导致成品树脂孔道中溶剂残留过高。
以上例举仅仅是对本发明的举例说明,并不构成对本发明的保护范围的限制,凡是与本发明相同或相似的设计均属于本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!